Advait
UcrainaIndice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE AДВЕЙТ (ADVATE)
Composizione:
Principio attivo: fattore della coagulazione VIII (octocog alfa);
1 flaconcino contiene:
250 UI* di fattore della coagulazione del sangue umano VIII ricombinante (DNA ricombinante)**, octocog alfa, che dopo ricostituzione corrisponde a circa 50 UI/ml di fattore della coagulazione del sangue umano VIII ricombinante, octocog alfa;
500 UI* di fattore della coagulazione del sangue umano VIII ricombinante (DNA ricombinante)**, octocog alfa, che dopo ricostituzione corrisponde a circa 100 UI/ml di fattore della coagulazione del sangue umano VIII ricombinante, octocog alfa;
1000 UI* di fattore della coagulazione del sangue umano VIII ricombinante (DNA ricombinante)**, octocog alfa, che dopo ricostituzione corrisponde a circa 200 UI/ml di fattore della coagulazione del sangue umano VIII ricombinante, octocog alfa;
1500 UI* di fattore della coagulazione del sangue umano VIII ricombinante (DNA ricombinante)**, octocog alfa, che dopo ricostituzione corrisponde a circa 300 UI/ml di fattore della coagulazione del sangue umano VIII ricombinante, octocog alfa;
2000 UI* di fattore della coagulazione del sangue umano VIII ricombinante (DNA ricombinante)**, octocog alfa, che dopo ricostituzione corrisponde a circa 400 UI/ml di fattore della coagulazione del sangue umano VIII ricombinante, octocog alfa;
3000 UI* di fattore della coagulazione del sangue umano VIII ricombinante (DNA ricombinante)**, octocog alfa, che dopo ricostituzione corrisponde a circa 600 UI/ml di fattore della coagulazione del sangue umano VIII ricombinante, octocog alfa;
Eccipienti: trealosio, istidina, idrossimetilaminometano, cloruro di sodio, cloruro di calcio, glutatione (ridotto), polisorbato 80, mannitolo.
* L'attività (in unità internazionali) è determinata mediante un test cromogenico quantitativo confrontato con uno standard interno, conforme allo standard dell'OMS. L'attività specifica è approssimativamente compresa tra 4000 e 10000 UI/mg di proteina.
** Il fattore della coagulazione del sangue umano VIII è prodotto mediante tecnologia del DNA ricombinante a partire da cellule ovariche del criceto cinese. Durante la coltivazione cellulare, nelle fasi di purificazione e nella formulazione finale non vengono aggiunte proteine (esogene) di origine umana o animale.
Forma farmaceutica. Polvere e solvente per soluzione per iniezione.
Principali caratteristiche fisico-chimiche: polvere granulare di colore bianco o quasi bianco; dopo ricostituzione, soluzione limpida, incolore, priva di particelle estranee.
Gruppo farmacoterapeutico. Sangue e organi ematopoietici. Agenti antiemorragici. Vitamina K e altri emostatici. Fattori della coagulazione. Fattore della coagulazione del sangue VIII.
Codice ATC B02B D02.
Proprietà farmacodinamiche.
Il complesso fattore VIII/fattore von Willebrand è costituito da due molecole (fattore VIII e fattore von Willebrand) con diverse funzioni fisiologiche. ADVATE contiene fattore VIII di coagulazione ricombinante umano (octocog alfa), una glicoproteina biologicamente equivalente alla glicoproteina fattore VIII presente nel plasma umano.
L'octocog alfa è una glicoproteina composta da 2332 amminoacidi con una massa molecolare approssimativa di 280 kDa. Dopo somministrazione per infusione a pazienti emofilici, l'octocog alfa si lega al fattore von Willebrand endogeno nel circolo sanguigno del paziente. Il fattore VIII attivato agisce come cofattore per il fattore IX attivato, accelerando la trasformazione del fattore X in fattore X attivato. Il fattore X attivato converte la protrombina in trombina. Successivamente, la trombina converte il fibrinogeno in fibrina, formando un coagulo di fibrina. L'emofilia A è un disturbo ereditario del sesso legato a un ridotto livello di attività del fattore VIII, che causa emorragie abbondanti nelle articolazioni, nei muscoli o negli organi interni, insorgenti spontaneamente o in seguito a traumi accidentali o chirurgici. I livelli plasmatici di fattore VIII aumentano grazie alla terapia sostitutiva, consentendo una correzione temporanea della carenza di fattore VIII e riducendo la tendenza all'emorragia.
Sono stati raccolti dati sull'induzione della tolleranza immunologica (ITI) in pazienti con inibitori al fattore VIII. In uno studio sottostudio 060103 che includeva bambini mai trattati precedentemente, le procedure ITI sono state documentate in 11 di questi pazienti. È stato effettuato un'analisi retrospettiva su 30 pazienti pediatrici in trattamento ITI (studio 060703). Nel registro non interventistico e prospettico (PASS-INT-004) sono stati documentati 44 pazienti pediatrici e adulti sottoposti a ITI, di cui 36 hanno completato la terapia ITI. I dati indicano che la tolleranza immunologica può essere raggiunta.
Nello studio 060201, 53 pazienti precedentemente trattati sono stati arruolati per confrontare due schemi di profilassi a lungo termine: un regime di dosaggio basato sulla farmacocinetica individuale (da 20 a 80 UI di fattore VIII per kg di peso corporeo ogni 72 ± 6 ore, n = 23) e un regime di dosaggio profilattico standard (da 20 a 40 UI/kg ogni 48 ± 6 ore, n = 30). Il regime basato sulla farmacocinetica (basato su una formula specifica) mira a mantenere livelli minimi di fattore VIII ≥ 1% durante l'intervallo di 72 ore tra le somministrazioni. I dati di questo studio indicano che entrambi gli schemi di dosaggio profilattico sono comparabili per quanto riguarda la riduzione della frequenza delle emorragie.
L'Agenzia Europea per i Medicinali ha esentato l'obbligo di presentare i risultati degli studi con il medicinale ADVATE in tutte le sottopopolazioni pediatriche con emofilia A (deficit congenito del fattore VIII): «Induzione della tolleranza immunologica (ITI) in pazienti con emofilia A (deficit congenito del fattore VIII) che sviluppano inibitori al fattore VIII» e «Trattamento e prevenzione delle emorragie in pazienti con emofilia A (deficit congenito del fattore VIII)» (si veda la sezione «Modalità di somministrazione e posologia» per informazioni sull'uso in pazienti pediatrici).
Proprietà farmacocinetiche.
Tutti gli studi farmacocinetici con il medicinale ADVATE sono stati condotti su pazienti precedentemente trattati affetti da emofilia A grave o moderatamente grave (livello iniziale di fattore VIII ≤ 2%). L'analisi dei campioni di plasma è stata effettuata in un laboratorio centrale mediante un test a un solo livello.
Complessivamente, 195 pazienti con emofilia A grave (livello iniziale di fattore VIII < 1%) hanno fornito parametri farmacocinetici inclusi nell'insieme di analisi farmacocinetiche previste dal protocollo. Le categorie di questi analisi per neonati (età da 1 mese a < 2 anni), bambini in età prescolare (da 2 a < 5 anni), bambini in età scolare (da 5 a < 12 anni), adolescenti (da 12 a < 18 anni) e adulti (età ≥ 18 anni) sono state utilizzate per riassumere i parametri farmacocinetici, dove l'età è stata definita come quella al momento dell'infusione PK.
Tabella 1
| Dati riassuntivi dei parametri farmacocinetici del medicinale Advait in pazienti con emofilia A grave (livello iniziale di fattore VIII < 1 %) |
|||||
| Parametro (valore medio ± deviazione standard) |
Lattanti (n = 5) |
Bambini in età prescolare (n = 30) |
Bambini in età scolare (n = 18) |
Adolescenti (n = 33) |
Adulti (n = 109) |
| AUC totale (UI * h/dl) |
1362,1 ± 311,8 |
1180,0 ± 432,7 |
1506,6 ± 530,0 |
1317,1 ± 438,6 |
1538,5 ± 519,1 |
| Risalita incrementale corretta a Cmax (UI/dl per UI/kg)a |
2,2 ± 0,6 |
1,8 ± 0,4 |
2,0 ± 0,5 |
2,1 ± 0,6 |
2,2 ± 0,6 |
| Emivita (ore) |
9,0 ± 1,5 |
9,6 ± 1,7 |
11,8 ± 3,8 |
12,1 ± 3,2 |
12,9 ± 4,3 |
| Concentrazione massima nel plasma dopo infusione (UI/dl) |
110,5 ± 30,2 |
90,8 ± 19,1 |
100,5 ± 25,6 |
107,6 ± 27,6 |
111,3 ± 27,1 |
| Emivita di eliminazione (ore) |
11,0 ± 2,8 |
12,0 ± 2,7 |
15,1 ± 4,7 |
15,0 ± 5,0 |
16,2 ± 6,1 |
| Volume di distribuzione a stato stazionario (dl/kg) |
0,4 ± 0,1 |
0,5 ± 0,1 |
0,5 ± 0,2 |
0,6 ± 0,2 |
0,5 ± 0,2 |
| Clearance (ml/kg * h) |
3,9 ± 0,9 |
4,8 ± 1,5 |
3,8 ± 1,5 |
4,1 ± 1,0 |
3,6 ± 1,2 |
a Calcolato come Cmax – livello basale del fattore VIII, diviso per la dose in UI/kg, dove Cmax è il valore massimo post-infusione del fattore VIII.
La sicurezza e l'efficacia emostatica del medicinale Advait nella popolazione pediatrica è simile a quella osservata negli adulti. Il recupero corretto e l'emivita terminale (t½) sono risultati approssimativamente il 20% più bassi nei bambini più piccoli (fino a 6 anni) rispetto agli adulti, fenomeno che può essere parzialmente attribuito all'aumento noto del volume plasmatico per chilogrammo di peso corporeo nei pazienti più giovani.
Ad oggi non sono disponibili dati sulla farmacocinetica del medicinale Advait nei pazienti precedentemente non trattati.
Caratteristiche cliniche.
Indicazioni.
Trattamento e profilassi delle emorragie in pazienti con emofilia A (deficit congenito del fattore VIII). Advait è indicato nei pazienti di tutte le fasce d'età.
Controindicazioni.
Ipersensibilità all'ingrediente attivo o a qualsiasi eccipiente elencato nella sezione «Composizione», oppure alle proteine di topo o di criceto.
Interazioni con altri medicinali ed altre forme di interazione.
Non sono stati condotti studi sulle interazioni per il medicinale Advait.
Caratteristiche particolari di utilizzo.
Tracciabilità
Per migliorare la tracciabilità dei medicinali biologici, si raccomanda di registrare il nome e il numero di lotto del prodotto somministrato.
Ipersensibilità
Durante l'uso del medicinale Advait sono state riportate reazioni di ipersensibilità, inclusa anafilassi. Il prodotto contiene proteine di topo e di criceto in tracce. In caso di comparsa di sintomi di ipersensibilità, ai pazienti deve essere raccomandato di interrompere immediatamente la somministrazione del medicinale e di consultare il medico. I pazienti devono essere informati sui segni precoci di reazioni di ipersensibilità, come orticaria, orticaria generalizzata, senso di costrizione al torace, sibili respiratori, ipotensione e anafilassi.
In caso di shock, devono essere avviate le consuete misure di emergenza.
Inibitori
La formazione di anticorpi neutralizzanti (inibitori) contro il fattore VIII è una complicanza nota nel trattamento dei pazienti con emofilia A. Tali inibitori sono solitamente immunoglobuline IgG dirette contro l'attività procoagulante del fattore VIII, espressi in unità Bethesda (UB) per 1 ml di plasma mediante test modificato. Il rischio di formazione di inibitori dipende dalla gravità della malattia e dal grado di esposizione al fattore VIII ed è massimo nei primi 20 giorni di trattamento. In rari casi, gli inibitori possono svilupparsi anche dopo oltre 100 giorni di terapia.
Sono stati osservati casi di recidiva di sviluppo di inibitori (a basso titolo) dopo oltre 100 giorni, in seguito al passaggio da un prodotto ricombinante di fattore VIII a un altro, in pazienti precedentemente trattati e con anamnesi positiva per sviluppo di inibitori. Pertanto, è necessario un monitoraggio attento dei pazienti per la comparsa di inibitori dopo il cambio di prodotto.
La rilevanza clinica della formazione di un inibitore dipende dal suo titolo. Gli inibitori a basso titolo, temporanei o persistentemente bassi, comportano un rischio minore di risposta clinica inadeguata rispetto agli inibitori ad alto titolo.
Tutti i pazienti trattati con fattore VIII della coagulazione devono essere attentamente monitorati per la comparsa di inibitori mediante osservazione clinica e test di laboratorio. Se i livelli attesi di attività del fattore VIII nel plasma non vengono raggiunti o se il sanguinamento non è controllato con la dose appropriata, si deve effettuare un test per la ricerca di inibitori del fattore VIII. Nei pazienti con alto titolo di inibitore, la terapia con fattore VIII può risultare inefficace e devono essere considerate alternative terapeutiche. Il trattamento di tali pazienti deve essere gestito da medici esperti nella gestione dell'emofilia e della formazione di inibitori del fattore VIII.
Complicanze legate alla cateterizzazione
Nel caso in cui sia necessario un dispositivo venoso centrale (CVAD), esiste il rischio di complicanze associate al CVAD, inclusi infezioni locali, batteriemia e trombosi nel sito di inserzione del catetere.
Fattori legati agli eccipienti
Sodio
Questo medicinale contiene 10 mg di sodio per flaconcino, pari allo 0,5% della dose giornaliera massima raccomandata dall'OMS di 2 g di sodio per l'adulto.
Si raccomanda vivamente di registrare ogni volta il nome e il numero di lotto del medicinale Advait somministrato al paziente, al fine di stabilire un collegamento tra lo stato del paziente e il lotto del medicinale.
Bambini
Le precauzioni e le misure di sicurezza nei bambini non differiscono da quelle degli adulti.
Uso durante la gravidanza e l’allattamento.
Non sono stati condotti studi sull’effetto del fattore VIII sulla riproduzione negli animali. A causa della bassa incidenza di emofilia A nelle donne, non esistono dati sull’uso del fattore VIII durante la gravidanza e l’allattamento. Pertanto, il fattore VIII deve essere utilizzato durante la gravidanza e l’allattamento solo in caso di chiare indicazioni.
Capacità di influenzare la capacità di guidare veicoli a motore o di usare macchinari.
Advait non ha alcun effetto sulla capacità di guidare veicoli a motore o di usare macchinari.
Modalità e dosi di somministrazione
Il trattamento deve essere iniziato sotto la supervisione di un medico esperto nella gestione dell'emofilia; devono inoltre essere disponibili misure di rianimazione in caso di anafilassi.
Dosaggio
Il dosaggio e la durata della terapia sostitutiva dipendono dal livello di carenza del fattore VIII, dalla sede e dall'entità dell'emorragia, nonché dallo stato clinico del paziente.
La quantità di unità del fattore VIII viene espressa in unità internazionali (UI), riferite allo standard OMS per i preparati di fattore VIII. L'attività del fattore VIII nel plasma viene espressa in percentuale (rispetto al plasma umano normale) oppure in UI (rispetto allo standard internazionale per il fattore VIII nel plasma sanguigno).
1 unità internazionale (UI) di attività del fattore VIII corrisponde alla quantità di fattore VIII presente in 1 ml di plasma umano normale.
Trattamento “on demand”
Il calcolo della dose richiesta di fattore VIII si basa sulla relazione empirica riscontrata: 1 UI di fattore VIII per kg di peso corporeo aumenta l'attività del fattore VIII di 2 UI/dl nel plasma. La dose necessaria viene determinata secondo la formula seguente:
- Dose richiesta (UI) = peso corporeo (kg) × incremento desiderato del fattore VIII (%) × 0,5
Nel caso di ulteriori episodi emorragici, l'attività del fattore VIII non deve scendere al di sotto di un determinato livello di attività plasmatica (in % rispetto al valore normale oppure in UI/dl) per il periodo appropriato. La seguente Tabella 2 può essere utilizzata come guida per la scelta della dose in caso di emorragie e interventi chirurgici.
Tabella 2
| Istruzione per la scelta della dose in caso di emorragia e interventi chirurgici |
||
| Grado di emorragia/tipo di intervento chirurgico |
Livello richiesto del fattore VIII (% o UI/dl) |
Frequenza di somministrazione (ore)/durata della terapia (giorni) |
| Emorragia |
||
| Emartrosi precoce, emorragia muscolare o emorragia orale. |
20–40 |
Ripetere le iniezioni ogni 12–24 ore (ogni 8–24 ore nei pazienti di età inferiore ai 6 anni) per almeno 1 giorno, fino alla cessazione dell’emorragia indicata dal dolore o fino al raggiungimento della guarigione. |
| Emartrosi avanzata, emorragia muscolare o ematoma. |
30–60 |
Ripetere le iniezioni ogni 12–24 ore (ogni 8–24 ore nei pazienti di età inferiore ai 6 anni) per 3–4 giorni o più a lungo, fino alla scomparsa del dolore o dell’incapacità funzionale. |
| Emorragia potenzialmente letale. |
60–100 |
Ripetere le iniezioni ogni 8–24 ore (ogni 6–12 ore nei pazienti di età inferiore ai 6 anni), fino alla scomparsa del pericolo per la vita. |
| Intervento chirurgico |
||
| Di lieve entità, incluso l’estrazione dentale |
30–60 |
Ogni 24 ore (ogni 12–24 ore nei pazienti di età inferiore ai 6 anni) per almeno 1 giorno, fino al raggiungimento della guarigione. |
| Di entità maggiore |
80–100 (prima e dopo l’intervento chirurgico) |
Ripetere le iniezioni ogni 8–24 ore (ogni 6–24 ore nei pazienti di età inferiore ai 6 anni) fino a una adeguata guarigione; successivamente continuare il trattamento per almeno 7 giorni per mantenere l’attività del fattore VIII tra il 30 % e il 60 % (UI/dl) |
La dose e la frequenza di somministrazione devono essere stabilite in base alla situazione clinica di ogni singolo paziente. In determinate circostanze (ad esempio, in presenza di un inibitore a basso titolo) può essere necessaria una dose superiore a quella calcolata secondo la formula.
Durante il trattamento è opportuno determinare i livelli di attività del fattore VIII nel plasma sanguigno e utilizzare tali valori per stabilire la dose necessaria e la frequenza delle iniezioni ripetute. In caso di interventi chirurgici maggiori, un controllo accurato della terapia sostitutiva mediante test quantitativo dell'attività del fattore VIII nel plasma sanguigno è indispensabile. Diversi pazienti possono presentare una risposta diversa al fattore VIII, raggiungendo differenti livelli di recupero in vivo e mostrando una diversa intensità di emivita.
Prevenzione
Per la profilassi a lungo termine delle emorragie nei pazienti con emofilia A grave, le dosi abituali sono di 20-40 UI di fattore VIII per kg di peso corporeo, con un intervallo di somministrazione da 2 a 3 giorni.
Modalità di somministrazione
ADVATE deve essere somministrato per via endovenosa. Se la somministrazione viene effettuata da una persona non medica, è necessaria un'adeguata preparazione.
La velocità di somministrazione deve essere tale da risultare confortevole per il paziente e non deve superare i 10 ml/min.
Dopo la ricostituzione, la soluzione deve essere limpida, incolore, priva di particelle estranee e con un pH compreso tra 6,7 e 7,3.
Istruzioni per la ricostituzione del medicinale prima della somministrazione
ADVATE deve essere somministrato per via endovenosa dopo la ricostituzione del prodotto.
Prima dell’uso, si deve effettuare un controllo visivo della soluzione ricostituita per verificare la presenza di particelle estranee e/o variazioni di colore.
Dopo la ricostituzione, la soluzione deve essere limpida, incolore e priva di inclusioni estranee. Non utilizzare soluzioni torbide o contenenti inclusioni.
- Per la somministrazione, utilizzare esclusivamente siringhe con attacco a camice di tipo Luer.
- Utilizzare entro tre ore dalla ricostituzione.
- La soluzione ricostituita non deve essere conservata in frigorifero.
Qualsiasi farmaco non utilizzato o rifiuti devono essere smaltiti in conformità ai requisiti locali.
Ricostituzione con il dispositivo BAXJECT II
- Per la ricostituzione utilizzare esclusivamente acqua sterile per preparazioni iniettabili e il dispositivo per la diluizione fornito nel kit.
- Non utilizzare se il dispositivo BAXJECT II, il suo sistema di barriera sterile o la confezione sono danneggiati o mostrano segni di deterioramento.
- È necessario rispettare le norme di asepsi.
- Se il medicinale è conservato in frigorifero, estrarre entrambe le fiale – quella contenente la polvere ADVATE e quella con il solvente – e lasciarle riscaldare a temperatura ambiente (circa 15-25 °C).
- Lavarsi accuratamente le mani con acqua calda e sapone.
- Rimuovere i tappi di protezione dalle fiale contenenti la polvere e il solvente.
- Disinfettare i setacci con tamponi alcolici. Posizionare le fiale su una superficie piana e pulita.
- Aprire la confezione del dispositivo BAXJECT II rimuovendo il coperchio di carta, senza toccare il dispositivo all’interno (fig. a). Non rimuovere il dispositivo dalla confezione. Non utilizzare se il dispositivo BAXJECT II, il suo sistema di barriera sterile o la confezione sono danneggiati o mostrano segni di deterioramento.
- Capovolgere la confezione e inserire la punta di plastica trasparente nel setaccio della fiala contenente il solvente. Afferrare la confezione per il bordo e rimuoverla dal dispositivo BAXJECT II (fig. b). Non rimuovere il cappuccio blu dal dispositivo BAXJECT II.
- Per la diluizione utilizzare esclusivamente acqua sterile per preparazioni iniettabili e il dispositivo per la diluizione fornito nel kit. Capovolgere il sistema con il dispositivo BAXJECT II collegato alla fiala del solvente in modo che la fiala si trovi sopra il dispositivo. Inserire la punta di plastica bianca nel setaccio della fiala contenente il medicinale ADVATE. Il vuoto attirerà il solvente nella fiala contenente ADVATE (fig. c).
- Mescolare delicatamente fino a completa dissoluzione del prodotto. Assicurarsi che ADVATE sia completamente disciolto, altrimenti non tutta la soluzione ricostituita passerà attraverso il filtro del dispositivo. Il prodotto si dissolve rapidamente (di solito in meno di 1 minuto). Dopo la ricostituzione, la soluzione deve essere limpida, incolore e priva di particelle estranee.
Fig. a Fig. b Fig. c
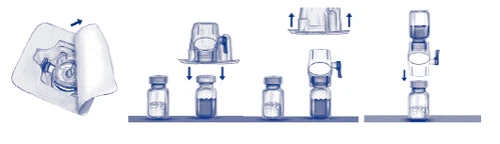
Somministrazione
Rispettare le norme di asepsi.
I medicinali per uso parenterale devono essere ispezionati visivamente per la presenza di particelle estranee immediatamente prima dell'uso, quando possibile in base al tipo di soluzione e di contenitore. Utilizzare solo soluzioni limpide e incolori.
- Rimuovere il cappuccio blu da BAXJECT II. Non aspirare aria nella siringa. Collegare la siringa al dispositivo BAXJECT II.
- Capovolgere il sistema (la fiala con la soluzione ricostituita deve trovarsi in alto). Aspirare lentamente la soluzione ricostituita nella siringa tirando delicatamente lo stantuffo.
- Staccare la siringa.
- Collegare l’ago a farfalla alla siringa. Somministrare per via endovenosa. La soluzione deve essere iniettata lentamente, con una velocità determinata dal comfort del paziente e non superiore a 10 ml al minuto. È necessario monitorare la frequenza cardiaca prima e durante la somministrazione di ADVATE. In caso di aumento significativo della frequenza cardiaca, ridurre la velocità di somministrazione o sospendere temporaneamente l’iniezione, il che di solito è sufficiente per una rapida scomparsa dei sintomi (vedere le sezioni «Precauzioni per l’uso» e «Effetti indesiderati»).
Dal punto di vista microbiologico, il medicinale deve essere somministrato immediatamente dopo la ricostituzione. Tuttavia, la stabilità chimica e fisica del medicinale dopo l’apertura della confezione è stata dimostrata per 3 ore a una temperatura di 25 °C.
Entro la data di scadenza, il prodotto può essere conservato a temperatura ambiente (non superiore a 25 °C) per un solo periodo non superiore a 6 mesi. La data di scadenza del periodo di 6 mesi di conservazione a temperatura ambiente deve essere indicata sull’imballaggio del medicinale. Il prodotto non può essere riportato in frigorifero per ulteriore conservazione.
Bambini
Nel trattamento "on demand", la posologia nei pazienti pediatrici (da 0 a 18 anni) non differisce da quella negli adulti. Ai pazienti di età inferiore a 6 anni, per la terapia profilattica, si somministrano da 20 a 50 UI di fattore VIII per kg di peso corporeo da 3 a 4 volte alla settimana.
Sovradosaggio.
Non sono stati riportati sintomi di sovradosaggio con questo medicinale.
Effetti indesiderati
Gli studi clinici sul medicinale ADVAYT hanno incluso 418 studi di somministrazione singola del medicinale ADVAYT, con manifestazione di 93 reazioni avverse. Gli effetti indesiderati frequenti comprendevano lo sviluppo di anticorpi neutralizzanti contro il fattore VIII (inibitori), cefalea e febbre.
Reazioni di ipersensibilità o reazioni allergiche (che possono includere angioedema, sensazione di calore e formicolio nel sito di infusione, brividi, iperemia, orticaria generalizzata, cefalea, orticaria, ipotensione, letargia, nausea, agitazione, tachicardia, oppressione toracica, pizzicore, vomito, sibili respiratori) sono state osservate raramente e in alcuni casi hanno avuto un andamento progressivo fino a provocare anafilassi acuta (incluso shock).
Nei pazienti che sviluppano anticorpi contro le proteine del topo e/o del criceto, possono verificarsi reazioni di ipersensibilità associate.
Nei pazienti con emofilia A che ricevono fattore VIII, incluso ADVAYT, possono formarsi anticorpi neutralizzanti (inibitori). Se si sviluppano tali inibitori, la risposta clinica risulterà inadeguata. In questi casi, si raccomanda di rivolgersi a un centro specializzato per il trattamento dell'emofilia.
Elenco delle reazioni avverse in forma di tabella
Nella tabella 3 riportata di seguito sono elencate le reazioni avverse al medicinale segnalate negli studi clinici e in segnalazioni spontanee, classificate per frequenza di insorgenza. La tabella segue la classificazione per sistemi e organi MedDRA (SOC) e termini preferenziali.
Le categorie di frequenza sono definite come segue: molto frequenti (≥ 1/10), frequenti (da ≥ 1/100 a < 1/10), non frequenti (da ≥ 1/1.000 a < 1/100), rari (da ≥ 1/10.000 a < 1/1.000), molto rari (< 1/10.000), non noti (non può essere stimato sulla base dei dati disponibili). All'interno di ciascuna categoria di frequenza, le reazioni avverse sono elencate in ordine decrescente di gravità.
Tabella 3
Frequenza di insorgenza delle reazioni avverse al medicinale segnalate negli studi clinici e in segnalazioni spontanee
| Sistema standardizzata di classi di organi/sistemi MedDRA |
Reazioni avverse |
Frequenzaa |
| Infezioni e infestazioni parassitarie |
Influenza |
Non comune |
| Laringite |
Non comune |
|
| Patologie del sistema emolinfopoietico |
Inibizione del fattore VIII |
Non comune (POPPL)d Molto comune (PBPL)d |
| Linfangite |
Non comune |
|
| Disturbi del sistema immunitario |
Reazione anafilattica |
Non noto |
| Iper-sensibilitàc |
Non noto |
|
| Disturbi del sistema nervoso |
Cefalea |
Comune |
| Vertigini |
Non comune |
|
| Peggioramento della memoria |
Non comune |
|
| Svenimento |
Non comune |
|
| Tremore |
Non comune |
|
| Emicrania |
Non comune |
|
| Dysgeusia |
Non comune |
|
| Disturbi della vista |
Infiammazione oculare |
Non comune |
| Patologie cardiache |
Palpitazioni |
Non comune |
| Disturbi vascolari |
Ematoma |
Non comune |
| Arrossamento del viso |
Non comune |
|
| Pallore |
Non comune |
|
| Patologie respiratorie, toraciche e mediastiniche |
Dispnea |
Non comune |
| Disturbi gastrointestinali |
Diarea |
Non comune |
| Dolore nell'addome superiore |
Non comune |
|
| Nausea |
Non comune |
|
| Vomito |
Non comune |
|
| Patologie della cute e del tessuto sottocutaneo |
Prurito |
Non comune |
| Eruzione cutanea |
Non comune |
|
| Iperidrosi |
Non comune |
|
| Orticaria |
Non comune |
|
| Patologie sistemiche e condizioni in relazione al sito di somministrazione |
Ipertermia |
Comune |
| Edema periferico |
Non comune |
|
| Dolore toracico |
Non comune |
|
| Disagio toracico |
Non comune |
|
| Brividi |
Non comune |
|
| Malessere |
Non comune |
|
| Ematoma nel sito di puntura vascolare |
Non comune |
|
| Stanchezza |
Non noto |
|
| Reazione nel sito di iniezione |
Non noto |
|
| Malessere generale |
Non noto |
|
| Esami di laboratorio |
Aumento del livello di monociti |
Non comune |
| Diminuzione del livello del fattore di coagulazione del sangue VIIIb |
Non comune |
|
| Diminuzione del livello di ematocrito |
Non comune |
|
| Deviazione dalla norma degli esiti di laboratorio |
Non comune |
|
| Lesioni, avvelenamenti e complicanze da procedure |
Complicanze post-procedura |
Non comune |
| Emorragia post-procedura |
Non comune |
|
| Reazione nel sito della procedura |
Non comune |
a Calcolato in base al numero totale di pazienti trattati con Advait (418).
b In un paziente, durante un'infusione continua di Advait dopo un intervento chirurgico (10-14 giorni dopo l'intervento), si è verificata una riduzione imprevista del livello del fattore della coagulazione VIII. Durante questo periodo è stato mantenuto un adeguato emostasi e i livelli plasmatici del fattore VIII e la velocità di eliminazione sono tornati ai valori normali il quindicesimo giorno dopo l'intervento. I test quantitativi per la ricerca di inibitori del fattore VIII, effettuati dopo la conclusione dell'infusione continua e dopo la fine dello studio, hanno dato esito negativo.
c La spiegazione relativa a questa reazione avversa al farmaco è riportata nella sezione seguente.
d La frequenza si basa su dati di studi con tutti i fattori VIII della coagulazione, inclusi pazienti con emofilia A grave. PPT: pazienti precedentemente trattati; PUNT: pazienti non precedentemente trattati.
Descrizione di reazioni avverse singole
Reazioni avverse ai residui del processo produttivo
In 229 pazienti sottoposti a trattamento è stata valutata la presenza di anticorpi contro le proteine cellulari del criceto cinese (CHO): 3 pazienti hanno mostrato un trend statisticamente significativo nei titoli, 4 hanno mostrato picchi persistenti o potenziali transitori e 1 paziente ha mostrato entrambi i segni, senza tuttavia manifestare sintomi clinici. Su 229 pazienti testati per la presenza di anticorpi IgG murini, 10 hanno mostrato un incremento statisticamente significativo, 2 hanno mostrato picchi persistenti o potenziali transitori e 1 paziente ha mostrato entrambi i segni. In 4 pazienti sono stati riportati singoli episodi di orticaria, prurito, eruzioni cutanee e un lieve aumento degli eosinofili in seguito a somministrazioni ripetute del medicinale.
Ipersensibilità
Le reazioni di tipo allergico includono anafilassi e si manifestano con capogiri, parestesie, eruzioni cutanee, arrossamento, gonfiore del viso, orticaria e prurito.
Bambini
Oltre alla formazione di anticorpi neutralizzanti (inibitori) nei bambini precedentemente non trattati e alle complicanze legate alla cateterizzazione, non sono state osservate differenze nelle reazioni avverse riscontrate negli studi clinici.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse sospette dopo l'autorizzazione del medicinale è di fondamentale importanza. Permette di monitorare il rapporto beneficio/rischio del medicinale. I professionisti sanitari e farmaceutici, nonché i pazienti o i loro rappresentanti legali, devono segnalare tutti i casi sospetti di reazioni avverse e di mancata efficacia del medicinale attraverso il sistema informativo automatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità.
2 anni.
Solvente (acqua per preparazioni iniettabili) – 5 anni.
Condizioni di conservazione.
Conservare a una temperatura compresa tra 2 e 8 °C. Non congelare! Conservare nell'imballaggio originale per proteggere dal contatto con la luce.
Conservare fuori dalla portata dei bambini.
Incompatibilità
Poiché non sono stati effettuati studi di compatibilità, questo medicinale non deve essere mescolato con altri farmaci o solventi.
Confezione.
1 flaconcino di polvere per soluzione iniettabile (250 UI, 500 UI, 1000 UI, 1500 UI, 2000 UI o 3000 UI) in combinazione con 1 flaconcino di solvente (5 ml di acqua per preparazioni iniettabili) e 1 dispositivo per la ricostituzione BAXJECT II in una confezione.
Categoria di distribuzione.
Sotto prescrizione medica.
Produttore.
Baxalta Belgium Manufacturing SA, Belgio / Baxalta Belgium Manufacturing SA, Belgium.
Indirizzo del produttore e sede operativa.
Boulevard Rene Branquart 80, Lessines, 7860, Belgio / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.