Vilate 500 UCI
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO VILATE 500 MO VILATE 1000 MO
Composición:
Principios activos: factor de coagulación de la sangre VIII, factor de von Willebrand;
1 frasco contiene 500 o 1000 MO de factor de coagulación de la sangre VIII y 500 MO o 1000 MO de factor de von Willebrand; contenido de proteína total ≤ 7,5 mg (500 MO) o ≤ 15 mg (1000 MO);
Excipientes: glicina, sacarosa, cloruro de sodio, citrato de sodio, cloruro de calcio.
Disolvente: agua para preparaciones inyectables con polisorbato 80 al 0,1 %.
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Principales propiedades físico-químicas:
Polvo: polvo o masa frágil de color blanco o amarillo pálido.
Disolvente: líquido incoloro transparente, sin olor. Sin partículas visibles.
Grupo farmacoterapéutico. Preparados antihemorrágicos. Factores de coagulación sanguínea. Factor de von Willebrand y factor de coagulación VIII en combinación. Código ATC B02B D06.
Propiedades farmacológicas.
Farmacodinámica.
Enfermedad de von Willebrand (VWD)
El factor de von Willebrand (proveniente del concentrado) es un componente normal del plasma sanguíneo humano y actúa de la misma manera que el factor de von Willebrand endógeno.
La administración del factor de von Willebrand permite corregir los trastornos hemostáticos que se manifiestan en pacientes con deficiencia del factor de von Willebrand, a dos niveles:
- El factor de von Willebrand restaura la adhesión de las plaquetas al subendotelio vascular en el sitio de la lesión vascular (ya que se une tanto al subendotelio vascular como a la membrana plaquetaria), asegurando así la hemostasia primaria, lo que se confirma mediante la reducción del tiempo de sangrado. Este efecto ocurre inmediatamente y, como se sabe, depende significativamente del grado de polimerización de la proteína;
- El factor de von Willebrand induce una corrección diferida del déficit asociado con el factor VIII. Tras la administración intravenosa, el factor de von Willebrand se une al factor VIII endógeno (normalmente producido por el paciente) y, al estabilizar este factor, evita su rápida degradación. Por ello, la administración de factor de von Willebrand puro (preparado de factor de von Willebrand con bajo contenido de factor VIII) restaura los niveles de factor VIII:C a valores normales como un efecto secundario tras la primera infusión. La administración de un preparado de factor de von Willebrand que contiene factor VIII restaura inmediatamente los niveles de factor VIII:C a valores normales tras la primera infusión.
Además de su función como proteína protectora del factor VIII, el factor de von Willebrand actúa como mediador en la adhesión plaquetaria a los sitios de lesión vascular y desempeña un papel en la agregación plaquetaria.
Hemofilia A
El complejo factor VIII / factor de von Willebrand está compuesto por dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas diferentes. Tras la administración al paciente con hemofilia, el factor VIII se une al factor de von Willebrand en la circulación sanguínea del paciente. El factor VIII activado (factor VIIIa) actúa como cofactor del factor IX activado (factor IXa), acelerando la transformación del factor X en factor X activado (factor Xa). El factor Xa convierte la protrombina en trombina. Posteriormente, la trombina transforma el fibrinógeno en fibrina, formándose así un coágulo.
La hemofilia A es un trastorno hereditario ligado al sexo del sistema de coagulación sanguínea, asociado con niveles reducidos de factor VIII:C (factor coagulante), lo que conduce a hemorragias severas en articulaciones, músculos o órganos internos, ya sea de forma espontánea o como consecuencia de un traumatismo accidental o quirúrgico. Mediante la terapia sustitutiva, los niveles plasmáticos de factor VIII aumentan, favoreciendo así una corrección temporal del déficit del factor y la corrección de la tendencia hemorrágica.
Cabe señalar que no es posible comparar directamente la tasa media anual de hemorragias (ABR/TMAH) observada en diferentes estudios clínicos ni en el caso del uso de diferentes concentrados de factor.
Además de su función como proteína protectora del factor VIII, el factor de von Willebrand actúa como mediador en la adhesión plaquetaria a los sitios de lesión vascular y desempeña un papel en la agregación plaquetaria.
Farmacocinética.
Enfermedad de von Willebrand (VWD)
El factor de von Willebrand (proveniente del concentrado) es un componente normal del plasma sanguíneo humano y actúa como el factor de von Willebrand endógeno.
Tabla 1
Resultados del metaanálisis de tres estudios farmacocinéticos con la participación de 24 pacientes con todos los tipos de VWD.
| Parámetro |
Todos los tipos de VWD |
VWD tipo 1 |
VWD tipo 2 |
VWD tipo 3 |
|||||||||||||||||
| N |
Valor medio |
DS |
Mín ∙ |
Máx ∙ |
N |
Valor medio |
DS |
Mín ∙ |
Máx ∙ |
N |
Valor medio |
DS |
Mín ∙ |
Máx ∙ |
N |
Valor medio |
DS |
Mín ∙ |
Máx ∙ |
||
| Recuperación (%/UI/kg) |
24 |
1,56 |
0,48 |
0,90 |
2,93 |
2 |
1,19 |
0,07 |
1,14 |
1,24 |
5 |
1,83 |
0,86 |
0,98 |
2,93 |
17 |
1,52 |
0,32 |
0,90 |
2,24 |
|
| AUC (0-inf) (g*%) |
23 |
1981 |
960 |
593 |
4831 |
2 |
2062 |
510 |
1701 |
2423 |
5 |
2971 |
1383 |
1511 |
4831 |
16 |
1662 |
622 |
593 |
2606 |
|
| T 1/2 (h) |
24 |
23,3 |
12,6 |
7,4 |
58,4 |
2 |
39,7 |
18,3 |
26,7 |
52,7 |
5 |
34,9 |
16 |
17,5 |
58,4 |
17 |
18 |
6,2 |
7,4 |
30,5 |
|
| MRT (h) |
24 |
33,1 |
19 |
10,1 |
89,7 |
2 |
53,6 |
25,9 |
35,3 |
71,9 |
5 |
53,5 |
24,6 |
27,8 |
89,7 |
17 |
24,7 |
8,5 |
10,1 |
37,7 |
|
| Depuración (ml/h/kg) |
24 |
3,29 |
1,67 |
0,91 |
7,41 |
2 |
2,66 |
0,85 |
2,06 |
3,27 |
5 |
1,95 |
1,02 |
0,91 |
3,31 |
17 |
3,76 |
1,69 |
1,83 |
7,41 |
|
AUC – área bajo la curva; MRT – tiempo medio de retención del medicamento en el organismo; SD – desviación estándar.
Hemofilia A
El factor VIII (procedente del concentrado) es un componente habitual del plasma sanguíneo humano y actúa como factor VIII endógeno. Tras la administración del medicamento, aproximadamente entre 2/3 y 3/4 del factor VIII permanece en circulación. El nivel de actividad del factor VIII:C alcanzado en el plasma sanguíneo representa entre el 80 % y el 120 % de la actividad esperada (prevista) del factor VIII.
La actividad del factor VIII disminuye mediante una eliminación exponencial bifásica. En la fase inicial, ocurre una distribución entre el compartimento intravascular y otros compartimentos (líquidos corporales), con un período de semivida en plasma de entre 3 y 6 horas. En la siguiente fase, más lenta, el período de semivida oscila entre 8 y 18 horas, con un promedio de 15 horas. Esto corresponde al período de semivida biológico real.
Tabla 2
Resultados de un estudio clínico con participación de 12 pacientes (análisis cromogénico, determinación mediante mediciones dobles).
| Parámetro |
Visita de nivel basal |
Visita a los 6 meses |
||
| Valor medio |
SD |
Valor medio |
SD |
|
| Recuperación (%/UI/kg) |
FVIII:C 2,27 |
1,20 |
FVIII:C 2,26 |
1,19 |
| AUCnorm (% × g/UI/kg) |
FVIII:C 31,3 |
7,31 |
FVIII:C 33,8 |
10,9 |
| Período de semieliminación (h) |
FVIII:C 11,2 |
2,85 |
FVIII:C 11,8 |
3,37 |
| MRT (h) |
FVIII:C 15,3 |
3,5 |
FVIII:C 16,3 |
4,6 |
| Depuración (ml/h/kg) |
FVIII:C 3,37 |
0,86 |
FVIII:C 3,24 |
1,04 |
AUC – área bajo la curva; MRT – tiempo medio de retención del fármaco en el organismo;
SD – desviación estándar.
Datos de estudios preclínicos de seguridad
El VWF y el factor VIII en VILATE son componentes normales del plasma sanguíneo humano y actúan como VWF/factor VIII endógeno.
Un estudio habitual de seguridad de estos componentes en animales de laboratorio no aportaría información útil adicional a la experiencia clínica existente; por lo tanto, no es necesario realizar dicho estudio.
Características clínicas.
Indicaciones.
Enfermedad de von Willebrand (VWD)
Prevención y tratamiento de hemorragias o sangrado durante intervenciones quirúrgicas en la enfermedad de von Willebrand (VWD), cuando el tratamiento con desmopresina (DDAVP) únicamente es ineficaz o está contraindicado.
Hemofilia A
Tratamiento y prevención de hemorragias en pacientes con hemofilia A (deficiencia congénita del factor de coagulación VIII).
Contraindicaciones.
Reacciones alérgicas a los principios activos o a cualquiera de los excipientes.
Interacción con otros medicamentos y otros tipos de interacciones.
No se conocen interacciones con otros medicamentos.
Características de uso.
Seguimiento
Para mejorar el seguimiento de medicamentos biológicos, debe registrarse claramente el nombre y el número de lote del medicamento administrado.
Hipersensibilidad
Al usar el medicamento VILATE pueden presentarse reacciones alérgicas. Además del factor VIII, el medicamento contiene trazas de proteínas humanas. Si aparecen síntomas de hipersensibilidad, se debe recomendar a los pacientes que interrumpan inmediatamente el uso del medicamento y consulten a su médico.
Se debe informar a los pacientes sobre los signos precoces de reacciones alérgicas, tales como: erupciones cutáneas, urticaria generalizada, dificultad respiratoria, disnea, hipotensión y anafilaxia.
En caso de shock, se debe aplicar un tratamiento anti-shock estándar.
Transmisión de agentes infecciosos
Las medidas habituales para prevenir infecciones derivadas del uso de medicamentos elaborados a partir de sangre o plasma humano incluyen la selección de donantes, el cribado de la sangre de donantes individuales y de lotes de plasma donado en busca de marcadores específicos de infección, así como la inclusión de etapas eficaces en el proceso de fabricación para la inactivación/eliminación de virus.
Sin embargo, cuando se administran medicamentos elaborados a partir de sangre o plasma humano, no puede excluirse por completo la posibilidad de transmisión de agentes infecciosos. Esto también incluye virus desconocidos o nuevos y otros microorganismos patógenos.
Se considera que las medidas adoptadas son eficaces frente a virus con envoltura, como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y el virus de la hepatitis C (VHC), así como frente al virus de la hepatitis A (VHA), que carece de envoltura. Las medidas adoptadas podrían tener una eficacia limitada frente a virus sin envoltura, como el parvovirus B19.
La infección por parvovirus B19 puede ser peligrosa para mujeres embarazadas (infección fetal) y para personas con inmunodeficiencia o eritropoyesis aumentada (por ejemplo, anemia hemolítica).
Se debe considerar la vacunación adecuada (hepatitis A y B) para pacientes que reciben de forma continua o repetida medicamentos de VWF/factor VIII elaborados a partir de plasma humano.
Se recomienda encarecidamente que cada vez que se administre el medicamento VILATE, el paciente registre el nombre y el número de lote del medicamento, con el fin de poder rastrear cualquier relación entre el estado del paciente y la administración del medicamento de un lote específico.
Enfermedad de von Willebrand (VWD)
Complicaciones tromboembólicas
Al usar un medicamento que contiene VWF con factor VIII, el médico que administra el tratamiento debe tener en cuenta que el tratamiento continuo puede provocar un aumento excesivo del factor VIII:C (factor coagulante VIII). En pacientes que reciben medicamentos con VWF que contienen factor VIII, se deben controlar los niveles plasmáticos de factor VIII:C para evitar niveles persistentemente elevados de factor VIII:C en plasma, ya que esto podría aumentar el riesgo de trombosis.
Existe riesgo de trombosis al usar medicamentos con VWF que contienen factor VIII, especialmente en pacientes con factores de riesgo clínicos o de laboratorio conocidos. Por lo tanto, se debe controlar el estado de los pacientes en grupos de riesgo en busca de signos precoces de trombosis. Se debe iniciar la profilaxis del tromboembolismo venoso según las recomendaciones vigentes.
Inhibidores
En pacientes con VWD, especialmente tipo 3, pueden desarrollarse anticuerpos neutralizantes (inhibidores) contra el VWF. Si no se alcanzan los niveles esperados de actividad de VWF:RCo en plasma o si la hemorragia no se controla con la dosis prescrita, se debe realizar el análisis correspondiente para determinar la presencia de inhibidores de VWF.
En pacientes con niveles altos de inhibidores, el tratamiento con VWF puede ser ineficaz, por lo que se deben considerar otras opciones terapéuticas. El tratamiento de estos pacientes debe ser supervisado por médicos con experiencia en el manejo de trastornos hemostáticos.
Hemofilia A
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII es una complicación conocida en el tratamiento de pacientes con hemofilia A.
Estos inhibidores suelen ser inmunoglobulinas de clase G (IgG) dirigidas contra la actividad procoagulante del factor VIII, cuya concentración se mide en unidades de Bethesda (BU) por 1 ml de plasma mediante un ensayo modificado. El riesgo de aparición de inhibidores está relacionado con la gravedad de la enfermedad y con la exposición al factor VIII. Este riesgo es mayor durante los primeros 50 días de exposición y permanece durante toda la vida, aunque sea infrecuente.
La relevancia clínica del desarrollo de inhibidores depende del título de los mismos: con títulos bajos, el riesgo de respuesta clínica insuficiente es menor que con títulos altos. En general, todos los pacientes tratados con medicamentos de coagulación que contienen factor VIII deben vigilarse cuidadosamente mediante observaciones clínicas y análisis de laboratorio adecuados para detectar la aparición de inhibidores.
Si no se alcanza el nivel esperado de actividad del factor VIII en plasma o si la hemorragia no se controla con la dosis prescrita, se debe realizar un estudio para detectar la presencia de inhibidores contra el factor VIII. En pacientes con niveles altos de inhibidores, el tratamiento con factor VIII puede ser ineficaz, por lo que se deben considerar otras opciones terapéuticas.
El manejo de estos pacientes debe realizarse bajo la supervisión de médicos con experiencia en el tratamiento de la hemofilia y en la formación de inhibidores contra el factor VIII.
Complicaciones cardiovasculares
En pacientes con factores de riesgo cardiovasculares preexistentes, la terapia sustitutiva con FVIII puede aumentar el riesgo de complicaciones cardiovasculares.
Complicaciones relacionadas con el catéter
Si se requiere el uso de un dispositivo de acceso venoso central (DAVC), se deben considerar los riesgos de complicaciones asociadas al DAVC, incluyendo infecciones locales, bacteriemia y trombosis en el sitio de inserción del catéter.
Este medicamento contiene hasta 58,7 mg de sodio por dosis de 500 UI de VWF y factor VIII/frasco y hasta 117,3 mg de sodio por dosis de 1000 UI de VWF y factor VIII/frasco, lo que equivale al 2,94 % y al 5,87 %, respectivamente, de la dosis diaria máxima recomendada por la OMS de 2 g de sodio para adultos.
Se debe tener en cuenta este contenido de sodio en pacientes sometidos a una dieta baja en sal.
Pacientes pediátricos
Las advertencias y precauciones especiales mencionadas se aplican tanto a adultos como a niños.
Uso durante el embarazo o la lactancia.
No se han realizado estudios de toxicidad reproductiva con VWF/factor VIII en animales.
Enfermedad de von Willebrand (VWD)
No existe experiencia en el tratamiento de mujeres embarazadas o en período de lactancia.
En caso de deficiencia de VWF, el medicamento VILATE debe administrarse a mujeres embarazadas o en período de lactancia solo si está claramente indicado, considerando que durante el parto existe un riesgo aumentado de hemorragia en estos pacientes.
Hemofilia A
Debido a la rareza de casos de hemofilia A en mujeres, no existe experiencia en el tratamiento durante el embarazo o la lactancia. Por lo tanto, VILATE debe usarse durante el embarazo y la lactancia solo si hay indicaciones claramente definidas.
Capacidad para conducir y usar máquinas.
VILATE no tiene ningún efecto sobre la capacidad para conducir vehículos ni para manejar maquinaria.
Vía de administración y dosis.
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de trastornos de la coagulación sanguínea. El medicamento en el frasco está indicado para uso único. Si queda algún contenido residual en el frasco después de su uso, este debe desecharse de acuerdo con los requisitos locales.
Enfermedad de von Willebrand (VWF)
La relación entre VWF:RCo y :C (ristocetina/cofactor del factor de von Willebrand y factor VIII coagulante) es aproximadamente de 1:1. En general, 1 UI/kg de peso corporal de VWF:RCo y FVIII:C aumenta la actividad de la proteína correspondiente en plasma en un 1,5–2 % respecto al valor normal. Habitualmente se requieren entre 20 y 50 UI de VILATE/kg de peso corporal para lograr un hemostasis adecuado. Esto provocará un aumento de VWF:RCo y FVIII:C en los pacientes de aproximadamente entre un 30 y 100 %.
Puede ser necesaria una dosis inicial de entre 50 y 80 UI de VILATE/kg de peso corporal, especialmente en pacientes con enfermedad de von Willebrand tipo 3, en quienes el mantenimiento de una actividad plasmática adecuada puede requerir dosis más altas que en pacientes con otros tipos de VWD.
Pacientes pediátricos
No hay suficientes datos disponibles para recomendar el uso de VILATE en niños menores de 6 años.
Profilaxis de hemorragias durante intervenciones quirúrgicas o trauma grave
Para la profilaxis de hemorragias durante intervenciones quirúrgicas, VILATE debe administrarse de 1 a 2 horas antes del inicio del procedimiento quirúrgico. Se deben alcanzar niveles de VWF:RCo ≥ 60 UI/dl (≥ 60 %) y niveles de FVIII:C ≥ 40 UI/dl (≥ 40 %).
La dosis adecuada debe repetirse cada 12–24 horas. La dosis y la duración del tratamiento dependen del estado clínico del paciente, el tipo y gravedad de la hemorragia, así como de los niveles de VWF:RCo y FVIII:C.
En pacientes que reciben medicamentos con VWF que contienen FVIII, debe controlarse el nivel de FVIII:C en plasma para evitar niveles persistentemente elevados, ya que esto podría aumentar el riesgo de trombosis, especialmente en pacientes con factores de riesgo clínicos o de laboratorio conocidos. Si se observan niveles excesivos de FVIII:C en plasma, se debe considerar la reducción de la dosis y/o el alargamiento del intervalo entre dosis, o el uso de un medicamento con VWF y bajo contenido de FVIII.
Profilaxis
Para la profilaxis prolongada de hemorragias en pacientes con VWD, se deben administrar dosis de 20–40 UI/kg de peso corporal 2 o 3 veces por semana. En algunos casos, como en hemorragias gastrointestinales, pueden requerirse dosis más altas.
Hemofilia A
Monitorización del tratamiento
Durante el curso del tratamiento, se recomienda determinar adecuadamente los niveles del factor VIII para establecer la dosis a administrar y la frecuencia de las infusiones repetidas. Diferentes pacientes pueden responder de forma distinta al tratamiento con factor VIII, mostrando distintos tiempos de semivida y recuperación. Los pacientes con peso corporal insuficiente o excesivo pueden requerir ajustes de la dosis basados en el peso. En particular, durante intervenciones quirúrgicas extensas, es obligatorio un control riguroso de la terapia sustitutiva mediante análisis de coagulación (actividad del factor VIII en plasma sanguíneo).
Dosis
La dosificación y duración de la terapia sustitutiva dependen del grado de gravedad del déficit del factor VIII, la localización y extensión de la hemorragia, así como del estado clínico del paciente.
La cantidad de unidades de factor VIII prescritas se expresa en unidades internacionales (UI), según el estándar vigente de la OMS para productos del factor VIII. La actividad del factor VIII en plasma se expresa bien en porcentaje (en relación con el plasma normal humano) o en unidades internacionales (en relación con el estándar internacional para FVIII en plasma).
Una unidad internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma normal humano.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en datos empíricos que indican que 1 UI de factor VIII:C/kg de peso corporal aumenta la actividad del factor en plasma entre un 1,5 y 2 % respecto al valor normal. La dosis requerida se determina mediante la siguiente fórmula:
UI necesarias = peso corporal (kg) × aumento deseado del factor VIII (%) (UI/dl) × 0,5 UI/kg
La cantidad y frecuencia de administración siempre deben ajustarse según la eficacia clínica en cada caso individual. En caso de complicaciones hemorrágicas, la actividad del factor VIII no debe descender por debajo del nivel de actividad plasmática establecido (en % del valor normal o en UI/dl) durante el período correspondiente.
La siguiente Tabla 3 puede utilizarse para determinar las dosis en caso de hemorragias y durante intervenciones quirúrgicas.
Tabla 3
Esquema de tratamiento para hemorragias e intervenciones quirúrgicas
| Grado de hemorragia/ Tipo de intervención quirúrgica |
Nivel necesario del factor VIII (%) (UI/dl) |
Frecuencia de dosis (horas)/ Duración del tratamiento (días) |
| Hemorragia |
||
| Hemartrosis inicial (hemorragia en la articulación), hemorragia muscular o hemorragia en la cavidad bucal |
20 – 40 |
Repetir cada 12 – 24 horas durante al menos 1 día, hasta que cese el sangrado (indicado por el dolor) o hasta lograr la recuperación. |
| Hemartrosis más extensa, hemorragia muscular o hematoma |
30 – 60 |
Repetir la inyección cada 12 – 24 horas durante 3 – 4 días o más, hasta que desaparezca el dolor y la limitación aguda del movimiento. |
| Hemorragias que ponen en peligro la vida |
60 – 100 |
Repetir la inyección cada 8 – 24 horas hasta que pase el riesgo vital. |
| Intervención quirúrgica |
||
| Pequeña intervención quirúrgica, incluyendo extracción dental |
30 – 60 |
Cada 24 horas, durante al menos 1 día, hasta lograr la recuperación. |
| Gran intervención quirúrgica |
80 – 100 (pre y postoperatorio) |
Repetir la inyección cada 8 – 24 horas hasta la completa cicatrización de la herida; posteriormente continuar el tratamiento durante al menos otros 7 días para mantener la actividad del factor VIII entre el 30% y el 60% (UI/dl). |
Prevención
Para la prevención a largo plazo de hemorragias en pacientes con hemofilia A grave, se deben administrar dosis habituales de 20 a 40 UI del factor VIII por kg de peso corporal cada 2-3 días. En algunos casos, especialmente en pacientes más jóvenes, podrían ser necesarios intervalos más cortos entre las administraciones o dosis más altas.
Infusión continua
Antes de una intervención quirúrgica, debe realizarse un análisis farmacocinético para estimar el aclaramiento. La velocidad inicial de administración del medicamento puede calcularse de la siguiente manera:
Velocidad de administración del medicamento (UI/kg/h) = aclaramiento (ml/kg/h) × nivel deseado en estado estacionario (UI/ml)
Después de las primeras 24 horas de infusión continua, el aclaramiento debe recalcularse diariamente utilizando la ecuación del estado estacionario, con el nivel medido y la velocidad de administración conocida.
Pacientes pediátricos
No hay datos suficientes para recomendar el uso de VILATE en niños menores de 6 años con hemofilia A.
Vía de administración
Administración intravenosa.
La velocidad de inyección o infusión no debe exceder los 2-3 ml por minuto.
Precauciones especiales para la eliminación y reciclaje
¡Debe leerse y seguirse cuidadosamente todas las instrucciones!
No debe utilizarse el medicamento después de la fecha de caducidad indicada en la etiqueta.
Durante el procedimiento descrito a continuación, debe mantenerse la esterilidad.
El medicamento diluido debe examinarse visualmente antes de la administración para detectar partículas visibles (sólidas) y cambios de color.
La solución debe ser transparente o ligeramente opalescente. No deben usarse soluciones turbias o con sedimentos.
La solución preparada debe usarse inmediatamente para evitar la contaminación microbiana.
Debe utilizarse únicamente el sistema de inyección suministrado. El uso de otro equipo de inyección o infusión puede provocar riesgos adicionales y conducir a una ineficacia del tratamiento.
Instrucciones para la preparación de la solución
- No usar el medicamento directamente desde el refrigerador. El diluyente y el polvo en los frascos cerrados deben alcanzar la temperatura ambiente.
- Retirar las tapas protectoras de ambos frascos y limpiar los tapones de goma con uno de los hisopos impregnados con alcohol suministrados en el envase.
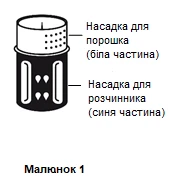
- El sistema de transferencia se muestra en la figura 1.
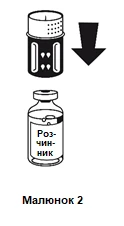
Colocar el frasco que contiene el diluyente sobre una superficie plana y nivelada y sujetarlo firmemente. Colocar el sistema de transferencia con la parte azul hacia arriba sobre el frasco que contiene el diluyente y presionar firmemente hacia abajo hasta que se escuche un clic (figuras 2 y 3).
No girar al fijar.
|
|
|
|
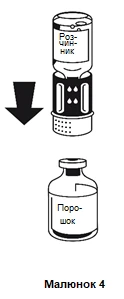
- Coloque el frasco que contiene el polvo sobre una superficie plana y estable, y sujételo firmemente. Tome el frasco con el disolvente junto con el sistema de transferencia fijado y inviértalo con la parte inferior hacia arriba. Coloque la parte blanca hacia arriba sobre el frasco que contiene el polvo y presione firmemente hacia abajo hasta que escuche un clic (figura 4). No gire al fijar. El disolvente pasará automáticamente al frasco que contiene el polvo.
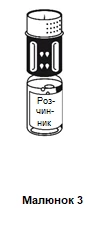
- Cuando ambos frascos aún estén unidos, gire suave y cuidadosamente el frasco que contiene el polvo hasta que el medicamento se disuelva completamente.
La disolución se produce en menos de 10 minutos a temperatura ambiente. Durante la preparación, puede formarse ligeramente espuma. Separe el sistema de transferencia en dos partes girándolo (figura 5). La formación de espuma desaparecerá.
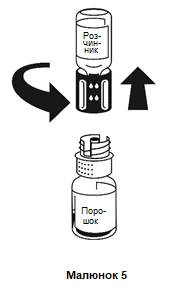
Deseche el frasco vacío que contiene el disolvente junto con la parte azul del sistema de transferencia.
Instrucciones para la administración de la inyección
Como medida de precaución, se debe comprobar la frecuencia del pulso del paciente antes y durante la inyección. Si se observa un aumento de la frecuencia del pulso, se debe reducir la velocidad de inyección o suspender temporalmente la administración.
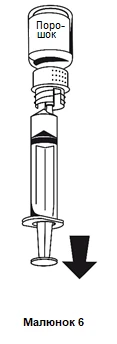
- 1. Acople la jeringa a la parte blanca del sistema de transferencia. Invierta el frasco y aspire la solución a la jeringa (figura 6).
La solución debe ser transparente o ligeramente opalescente.
Tan pronto como se haya aspirado la solución, sujetando firmemente la jeringa por el cilindro, desacople la jeringa del sistema de transferencia (figura 7).
|
|
|
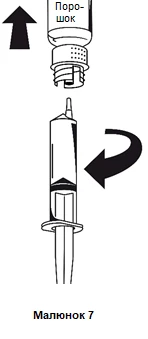
Se debe desechar el frasco vacío junto con la parte blanca del sistema de transferencia.
- 2. Desinfectar el lugar elegido para la inyección con una de las compresas impregnadas de alcohol suministradas en el envase.
- Conectar el sistema de administración suministrado a la jeringa.
- Introducir la aguja de inyección en la vena deseada. Si se utiliza un torniquete (cinta de presión) para visualizar mejor la vena, este debe aflojarse antes de comenzar la administración de Vilate.
La sangre no debe entrar en la jeringa debido al riesgo de formación de coágulos de fibrina.
- Administrar la solución en la vena muy lentamente, a una velocidad de 2–3 ml por minuto.
Si se utilizan más de un frasco de polvo de Vilate por administración, se puede volver a usar la misma aguja de inyección y jeringa. ¡El sistema de transferencia está diseñado únicamente para uso único (usar solo una vez)!
Cualquier medicamento no utilizado o residuos deben desecharse de acuerdo con los requisitos locales.
Niños.
Enfermedad de von Willebrand (VWD)
No hay suficientes datos para recomendar el uso de VILATE en niños menores de 6 años.
Hemofilia A
No hay suficientes datos para recomendar el uso de VILATE en niños menores de 6 años con hemofilia A.
Sobredosis.
No se han notificado síntomas de sobredosis de FVIII o VWF en humanos. En caso de sobredosis significativa, podrían presentarse complicaciones tromboembólicas.
Reacciones adversas.
Resumen breve del perfil de seguridad
Se han observado reacciones de hipersensibilidad o alérgicas (que pueden incluir: angioedema, sensación de ardor y picazón en el lugar de la inyección, escalofríos, hiperemia (rubor), urticaria generalizada, enrojecimiento, prurito, erupción cutánea, cefalea, urticaria, hipotensión, somnolencia, náuseas, excitación, taquicardia, dificultad respiratoria, disnea, hormigueo, vómitos, sibilancias/respiración sibilante) raramente, y en algunos casos pueden progresar hasta una anafilaxia grave (incluyendo shock).
Enfermedad de von Willebrand (VWD)
En pacientes con VWD, especialmente tipo 3, pueden desarrollarse muy raramente anticuerpos neutralizantes contra el VWF. Si aparecen tales inhibidores, se manifestarán mediante una respuesta clínica inadecuada. Estos anticuerpos pueden presentarse simultáneamente con reacciones anafilácticas. Por lo tanto, los pacientes con reacción anafiláctica deben ser evaluados para detectar la presencia de inhibidores.
En todos los casos con reacción anafiláctica se recomienda consultar a un centro especializado en el tratamiento de la hemofilia.
Existe un riesgo de trombosis, especialmente en pacientes con factores clínicos o de laboratorio conocidos de riesgo. Se debe iniciar la profilaxis contra la trombosis venosa de acuerdo con las recomendaciones vigentes.
En pacientes que reciben tratamientos con VWF que contienen FVIII, los niveles persistentemente elevados de FVIII:C en plasma sanguíneo pueden aumentar el riesgo de formación de trombos.
Hemofilia A
En pacientes con hemofilia A que han recibido tratamiento con factor VIII y el medicamento VILATE, pueden desarrollarse anticuerpos neutralizantes (inhibidores), véase la sección «Propiedades farmacodinámicas». Si aparecen tales inhibidores, la condición se manifestará como una respuesta clínica insuficiente. En tales casos, se recomienda consultar a un centro especializado en el tratamiento de la hemofilia.
Lista tabulada de reacciones adversas
La frecuencia de aparición de reacciones adversas se clasifica según las siguientes categorías: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 a < 1/10); poco frecuentes (≥ 1/1.000 a < 1/100); raras (≥ 1/10.000 a < 1/1.000); muy raras (< 1/10.000); frecuencia desconocida (no puede determinarse con los datos disponibles).
En la tabla 4 se indican las reacciones adversas observadas en estudios clínicos, estudios de seguridad poscomercialización, así como aquellas conocidas a partir de otras fuentes poscomercialización. Las reacciones adversas se presentan por clases de sistemas y órganos (SOC) de MedDRA, utilizando términos preferentes (PT) y clasificadas por frecuencia.
Tabla 4
| Clase de sistema de órganos (SOC/CSO) de MedDRA |
Reacción adversa |
Frecuencia |
| Alteraciones del sistema inmunitario |
Hipersensibilidad Anafilaxia |
No frecuente Muy raro |
| Alteraciones generales y en el lugar de administración |
Fiebre Dolor en el pecho |
No frecuente Frecuencia desconocida |
| Alteraciones de la sangre y del sistema linfático |
Inhibidores del factor VIII Inhibidores del factor de von Willebrand |
No frecuente (PTPs)* Muy frecuente (PUPs) Muy raro |
| Alteraciones del sistema respiratorio, del tórax y de la pleura |
Tos |
Frecuencia desconocida |
| Alteraciones del sistema nervioso |
Vertigo |
Frecuencia desconocida |
| Alteraciones del aparato gastrointestinal |
Dolor abdominal |
Frecuencia desconocida |
| Alteraciones del músculo esquelético, del tejido conjuntivo y del sistema osteoarticular |
Dolor de espalda |
Frecuencia desconocida |
* La frecuencia se basa en estudios con todos los medicamentos FVIII realizados en pacientes con hemofilia A grave. PTP: pacientes previamente tratados; PUP: pacientes previamente no tratados.
Descripción de reacciones adversas individuales
Para información sobre reacciones adversas individuales, véase la sección «Precauciones de uso».
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite continuar con el seguimiento de la relación beneficio/riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar cualquier caso sospechoso de reacción adversa o falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en el enlace: https://aisf.dec.gov.ua.
Período de validez
Polvo para solución inyectable: 3 años.
La estabilidad de la solución reconstituida se mantiene durante 4 horas a temperatura ambiente (no superior a 25 °C). Sin embargo, para evitar la contaminación por microorganismos, la solución reconstituida debe usarse inmediatamente.
Solvente: conservar durante 4 años.
El solvente debe conservarse durante 4 años a una temperatura de entre 2 y 8 °C, en un lugar protegido de la luz.
Durante este período, el solvente puede mantenerse hasta 6 meses a una temperatura no superior a 25 °C; en este caso, el período de validez finaliza a los 6 meses.
Condiciones de almacenamiento
Conservar a una temperatura de entre 2 y 8 °C. No congelar.
Conservar el frasco en su envase de cartón para protegerlo de la luz.
Mantener fuera del alcance de los niños.
Durante el período de validez, el medicamento puede almacenarse a temperatura ambiente (no superior a 25 °C) durante un máximo de 2 meses. En este caso, el período de validez finaliza a los 2 meses desde la primera extracción del medicamento del refrigerador.
El paciente debe indicar la nueva fecha de caducidad en el envase exterior de cartón.
La solución reconstituida está destinada para uso único. Cualquier solución restante en el frasco debe desecharse.
Incompatibilidades
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos ni administrarse simultáneamente con otro medicamento intravenoso en el mismo sistema de infusión. Debe utilizarse únicamente el sistema de inyección/infusión suministrado, ya que el tratamiento podría no ser eficaz debido a la adsorción del factor VIII/factor de von Willebrand por la superficie interna de ciertos dispositivos de infusión.
Envase
Caja de cartón n.º 1: 1 frasco con polvo para preparar solución inyectable (500 UI) e instrucciones de uso.
Caja de cartón n.º 2: 1 frasco con solvente (agua para inyección con polisorbato 80 al 0,1 %), 5 ml, junto con un kit para administración intravenosa y 2 torundas impregnadas con alcohol.
El kit para administración intravenosa incluye: 1 jeringa desechable, 1 dispositivo de transferencia, 1 sistema de infusión.
La caja de cartón n.º 1 y la caja de cartón n.º 2 están unidas entre sí mediante una película plástica.
Caja de cartón n.º 1: 1 frasco con polvo para preparar solución inyectable (1000 UI) e instrucciones de uso.
Caja de cartón n.º 2: 1 frasco con solvente (agua para inyección con polisorbato 80 al 0,1 %), 10 ml, junto con un kit para administración intravenosa y 2 torundas impregnadas con alcohol.
El kit para administración intravenosa incluye: 1 jeringa desechable, 1 dispositivo de transferencia, 1 sistema de infusión.
La caja de cartón n.º 1 y la caja de cartón n.º 2 están unidas entre sí mediante una película plástica.
Categoría de dispensación: Bajo receta médica.
Fabricante: Octapharma Pharmazeutika Produktionsges.m.b.H.
Domicilio del fabricante y dirección del lugar de actividad.
Oberlaaer Straße 235, 1100 Viena, Austria.