Sumamed® Forte
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO SUMAMED® FORTE (SUMAMED® FORTE)
Composición:
Principio activo: azitromicina;
1 dosis (5 ml) de suspensión contiene 200 mg de azitromicina en forma de dihidrato de azitromicina;
Excipientes: sacarosa, fosfato de sodio, hidroxipropilcelulosa, goma xantana, dióxido de silicio coloidal anhidro,
saborizantes y/o colorantes:
saborizante de banana, saborizante de cereza, saborizante de vainilla
o
dióxido de titanio (E 171), saborizante de banana, saborizante de vainilla,
o
dióxido de titanio (E 171), saborizante de fresa,
o
dióxido de titanio (E 171), saborizante de frambuesa.
Forma farmacéutica. Polvo para suspensión oral.
Propiedades físicas y químicas principales: polvo de color blanco o blanco amarillento con aroma característico a cereza y banana, o banana, o fresa, o frambuesa.
La suspensión reconstituida es una suspensión homogénea de color blanco amarillento con aroma característico a cereza y banana, o banana, o fresa, o frambuesa.
Grupo farmacoterapéutico. Agentes antibacterianos para uso sistémico. Macrólidos, lincominas y estreptograminas. Azitromicina. Código ATC J01FA10.
Propiedades farmacológicas.
Farmacodinamia.
La azitromicina es un antibiótico macrólido que pertenece al grupo de los azálidos. La molécula se forma mediante la introducción de un átomo de nitrógeno en el anillo lactónico de la eritromicina A. El mecanismo de acción de la azitromicina consiste en la inhibición de la síntesis de proteínas bacterianas mediante unión a la subunidad 50S de los ribosomas e inhibición de la translocación de péptidos.
Mecanismo de resistencia.
Existe una resistencia cruzada completa en Streptococcus pneumoniae, estreptococo beta-hemolítico del grupo A, Enterococcus faecalis y Staphylococcus aureus, incluyendo el Staphylococcus aureus resistente a la meticilina (MRSA), frente a la eritromicina, la azitromicina, otros macrólidos y los lincosamidas.
La prevalencia de resistencia adquirida puede variar según la región y el tiempo, por lo que se requiere información local sobre la resistencia, especialmente en el tratamiento de infecciones graves. Si es necesario, se debe consultar a un especialista, particularmente cuando la prevalencia local de resistencia sea tal que la eficacia del medicamento para tratar al menos algunos tipos de infecciones sea dudosa.
Espectro de actividad antimicrobiana de la azitromicina
| Especies habitualmente sensibles |
| Bacterias aerobias grampositivas |
| Staphylococcus aureus sensible a la meticilina |
| Streptococcus pneumoniae sensible a la penicilina |
| Streptococcus pyogenes |
| Bacterias aerobias gramnegativas |
| Haemophilus influenzae Haemophilus parainfluenzae |
| Legionella pneumophila |
| Moraxella catarrhalis |
| Pasteurella multocida |
| Bacterias anaerobias |
| Clostridium perfringens |
| Fusobacterium spp. |
| Prevotella spp. |
| Porphyriomonas spp. |
| Otros microorganismos |
| Chlamydia trachomatis Chlamydia pneumoniae Mycoplasma pneumoniae |
| Especies para las que la resistencia adquirida puede ser un problema |
| Bacterias aerobias grampositivas |
| Streptococcus pneumoniae con sensibilidad intermedia a la penicilina y resistente a la penicilina |
| Organismos intrínsecamente resistentes |
| Bacterias aerobias grampositivas |
| Enterococcus faecalis |
| Estafilococos MRSA, MRSE* |
| Bacterias anaerobias |
| Grupo de bacteroides Bacteroides fragilis |
*Metici-lina* -resistente Staphylococcus aureus tiene una resistencia adquirida muy alta a los macrólidos y se menciona aquí debido a la rara sensibilidad a la azitromicina.
Farmacocinética.
La biodisponibilidad tras la administración oral es aproximadamente del 37 %. La concentración máxima en suero sanguíneo se alcanza entre 2 y 3 horas tras la ingestión del fármaco.
Tras la administración oral, la azitromicina se distribuye por todo el organismo. En estudios farmacocinéticos se ha demostrado que la concentración de azitromicina en los tejidos es significativamente más alta (hasta 50 veces) que en el plasma sanguíneo, lo que indica una fuerte unión del fármaco a los tejidos.
La unión a las proteínas plasmáticas varía según las concentraciones plasmáticas y oscila entre el 12 % a 0,5 µg/ml y el 52 % a 0,05 µg/ml en suero sanguíneo. El volumen aparente de distribución en estado de equilibrio (VVss) es de 31,1 l/kg.
El período final de semivida plasmática refleja completamente el período de semivida en los tejidos durante 2 a 4 días.
Aproximadamente el 12 % de la dosis intravenosa de azitromicina se excreta sin cambios en la orina durante los tres días siguientes. Se han detectado concentraciones particularmente altas de azitromicina inalterada en la bilis humana. También se han identificado diez metabolitos en la bilis, formados por desmetilación N y O, hidroxilación de los anillos de desosamina y aglicona, y escisión del conjugado de cladinosa. La comparación de los resultados de cromatografía líquida y análisis microbiológicos mostró que los metabolitos de la azitromicina no son microbiológicamente activos.
Características clínicas.
Indicaciones.
Infecciones causadas por microorganismos sensibles a la azitromicina:
- Infecciones de oído, nariz y garganta (faringitis/tonsilitis bacteriana, sinusitis, otitis media);
- Infecciones del tracto respiratorio (bronquitis bacteriano, neumonía adquirida en la comunidad);
- Infecciones de la piel y tejidos blandos: eritema migrans (estadio inicial de la enfermedad de Lyme), escarificación, impétigo, piodermitis secundarias.
Contraindicaciones.
Hipersensibilidad al azitromicina, eritromicina, a cualquier antibiótico macrólido o cetólido, así como a cualquiera de los demás componentes del medicamento.
Interacción con otros medicamentos y otros tipos de interacciones.
Antiácidos. En estudios sobre el efecto de la administración concomitante de antiácidos sobre la farmacocinética de la azitromicina, no se observaron cambios generales en la biodisponibilidad, aunque la concentración plasmática máxima de azitromicina disminuyó aproximadamente un 25 %. No se debe tomar azitromicina y antiácidos simultáneamente.
Cetirizina. En voluntarios sanos, la administración concomitante de azitromicina durante 5 días con cetirizina 20 mg en estado de equilibrio no mostró interacción farmacocinética ni cambios significativos en el intervalo QT.
Didanosina. En seis voluntarios VIH positivos, la administración concomitante de dosis diarias de 1200 mg de azitromicina con 400 mg de didanosina al día no mostró efecto sobre la farmacocinética de la didanosina en estado de equilibrio en comparación con placebo.
Digoxina y colchicina. Se han notificado casos de administración concomitante de antibióticos macrólidos, incluyendo azitromicina, y sustratos de la glucoproteína P, como digoxina y colchicina, que provocan un aumento en los niveles séricos de estos sustratos. Por lo tanto, al administrar conjuntamente azitromicina y un sustrato de la glucoproteína P como la digoxina, debe considerarse la posibilidad de un aumento en la concentración sérica del sustrato.
Zidovudina. Dosis únicas de 1000 mg y dosis múltiples de 1200 mg o 600 mg de azitromicina tuvieron un efecto insignificante sobre la farmacocinética plasmática o la excreción urinaria de zidovudina o sus metabolitos glucurónidos. Sin embargo, la administración de azitromicina aumentó las concentraciones de zidovudina fosforilada, metabolito clínicamente activo, en los mononucleares de la circulación periférica. La relevancia clínica de estos datos es incierta, pero podría ser beneficiosa para los pacientes.
La azitromicina no tiene interacción significativa con el sistema hepático del citocromo P450. Se considera que el medicamento no presenta interacción farmacocinética propia de la eritromicina y otros macrólidos. La azitromicina no induce ni inactiva el citocromo P450 hepático mediante complejos metabolitos-citocromo.
Ergotamina. Debido a la posibilidad teórica de ergotismo, no se recomienda la administración concomitante de azitromicina con derivados del ergot.
Se han realizado estudios farmacocinéticos sobre la administración conjunta de azitromicina y los siguientes medicamentos cuyo metabolismo depende en gran medida del citocromo P450.
Atorvastatina. La administración concomitante de atorvastatina (10 mg/día) y azitromicina (500 mg/día) no provocó cambios en las concentraciones plasmáticas de atorvastatina (basado en el análisis de inhibición de la HMG-CoA reductasa). Sin embargo, durante el período poscomercialización se han registrado casos de rabdomiólisis en pacientes que tomaban azitromicina junto con estatinas.
Carbamazepina. En un estudio de interacción farmacocinética en voluntarios sanos, la azitromicina no mostró efecto significativo sobre los niveles plasmáticos de carbamazepina ni sobre sus metabolitos activos.
Cimetidina. En un estudio farmacocinético sobre el efecto de una dosis única de cimetidina administrada 2 horas antes de la azitromicina sobre la farmacocinética de esta última, no se observaron cambios en la farmacocinética de la azitromicina.
Anticoagulantes orales tipo cumarina. En un estudio de interacción farmacocinética, la azitromicina no alteró el efecto anticoagulante de una dosis única de 15 mg de warfarina administrada a voluntarios sanos. Durante el período poscomercialización se han recibido informes sobre potenciación del efecto anticoagulante tras la administración concomitante de azitromicina y anticoagulantes orales tipo cumarina. Aunque no se ha establecido un vínculo causal, debe considerarse la necesidad de realizar un monitoreo frecuente del tiempo de protrombina cuando se administre azitromicina a pacientes que reciben anticoagulantes orales tipo cumarina.
Ciclosporina. En un estudio farmacocinético con voluntarios sanos que recibieron una dosis oral diaria de azitromicina de 500 mg durante 3 días, seguida de una dosis oral única de ciclosporina de 10 mg/kg, se observó un aumento significativo de la Cmáx y del AUC0-5 de ciclosporina. Por lo tanto, debe tenerse precaución al administrar estos medicamentos conjuntamente. Si la administración concomitante es necesaria, deben monitorizarse los niveles de ciclosporina y ajustarse la dosis en consecuencia.
Efavirenz. La administración concomitante de una dosis única de azitromicina de 600 mg y 400 mg de efavirenz diarios durante 7 días no provocó ninguna interacción farmacocinética clínicamente relevante.
Fluconazol. La administración concomitante de una dosis única de azitromicina de 1200 mg no altera la farmacocinética de una dosis única de fluconazol de 800 mg. La exposición total y el período de semivida de la azitromicina no se modificaron con la administración concomitante de fluconazol, aunque se observó una disminución clínicamente insignificante de la Cmáx (18 %) de azitromicina.
Indinavir. La administración concomitante de una dosis única de azitromicina de 1200 mg no produce un efecto estadísticamente significativo sobre la farmacocinética del indinavir administrado a dosis de 800 mg tres veces al día durante 5 días.
Metilprednisolona. En un estudio de interacción farmacocinética en voluntarios sanos, la azitromicina no afectó significativamente la farmacocinética de la metilprednisolona.
Midazolam. En voluntarios sanos, la administración concomitante de azitromicina 500 mg/día durante 3 días no provocó cambios clínicamente significativos en la farmacocinética y farmacodinámica del midazolam administrado como dosis única de 15 mg.
Nelfinavir. La administración concomitante de azitromicina (1200 mg) y nelfinavir en estado de equilibrio (750 mg tres veces al día) provoca un aumento en la concentración de azitromicina. No se observaron efectos adversos clínicamente significativos, por lo tanto no es necesaria la corrección de la dosis.
Rifabutina. La administración concomitante de azitromicina y rifabutina no afectó las concentraciones séricas de estos medicamentos. Se observó neutropenia en sujetos que tomaban azitromicina y rifabutina simultáneamente. Aunque la neutropenia estuvo asociada con el uso de rifabutina, no se estableció un vínculo causal con la administración concomitante de azitromicina.
Sildenafil. En voluntarios sanos del sexo masculino no se obtuvieron evidencias del efecto de la azitromicina (500 mg/día durante 3 días) sobre los valores de AUC y Cmáx de sildenafil o de su principal metabolito circulante.
Terfenadina. En estudios farmacocinéticos no se notificó interacción entre azitromicina y terfenadina. En algunos casos no puede descartarse completamente la posibilidad de dicha interacción; sin embargo, no existen datos específicos sobre su presencia.
Teofilina. No existen datos sobre interacción farmacocinética clínicamente significativa con la administración concomitante de azitromicina y teofilina en voluntarios sanos.
Triazolam. La administración concomitante en voluntarios sanos de azitromicina (500 mg el primer día y 250 mg el segundo día) con 0,125 mg de triazolam no afectó significativamente ninguno de los parámetros farmacocinéticos del triazolam en comparación con el uso de triazolam y placebo.
Trimetoprim/sulfametoxazol. La administración concomitante durante 7 días de trimetoprim/sulfametoxazol en doble concentración (160 mg/800 mg) con azitromicina de 1200 mg en el séptimo día no mostró efecto significativo sobre las concentraciones máximas, la exposición total ni la excreción urinaria de trimetoprim o sulfametoxazol. Los valores de concentración de azitromicina en suero fueron similares a los observados en otros estudios.
Hidroxicloroquina. La azitromicina debe administrarse con precaución a pacientes que reciben medicamentos que prolongan el intervalo QT y pueden provocar arritmias cardíacas, como la hidroxicloroquina.
Características de uso.
Al igual que con la eritromicina y otros antibióticos macrólidos, se han notificado reacciones alérgicas graves aisladas, incluyendo angioedema y anafilaxia (en casos aislados con resultado fatal), reacciones dermatológicas, incluyendo pustulosis exantemática aguda generalizada. Algunas de estas reacciones provocadas por azitromicina provocaron síntomas recurrentes y requirieron observación y tratamiento más prolongados.
Dado que el hígado es la vía principal de eliminación de la azitromicina, debe tenerse precaución al administrar azitromicina a pacientes con enfermedad hepática grave. Se han notificado casos de hepatitis fulminante que provocan insuficiencia hepática potencialmente mortal durante el tratamiento con azitromicina. Posiblemente algunos pacientes tenían antecedentes de enfermedad hepática o estaban tomando otros medicamentos hepatotóxicos.
Se deben realizar análisis/pruebas de función hepática si aparecen signos y síntomas de disfunción hepática, como astenia que progresa rápidamente y se asocia con ictericia, orina oscura, tendencia al sangrado o encefalopatía hepática. Si se detecta alteración de la función hepática, debe interrumpirse el tratamiento con azitromicina.
En pacientes que toman derivados del cornezuelo, la administración concomitante de ciertos antibióticos macrólidos favorece el desarrollo rápido de ergotismo. No existen datos sobre la posibilidad de interacción entre derivados del cornezuelo y azitromicina. Sin embargo, debido a la posibilidad teórica de ergotismo, no se debe administrar azitromicina simultáneamente con derivados del cornezuelo.
Como ocurre con otros antibióticos, se recomienda observar signos de superinfección provocada por microorganismos no sensibles, incluyendo hongos.
Durante el tratamiento con casi todos los agentes antibacterianos, incluyendo la azitromicina, se han notificado casos de diarrea asociada con Clostridium difficile (CDAD), cuya gravedad varía desde diarrea leve hasta colitis con resultado fatal. El tratamiento con antibacterianos altera la flora normal del intestino grueso, lo que conduce al crecimiento excesivo de C. difficile.
C. difficile produce toxinas A y B, que contribuyen al desarrollo de CDAD. Las cepas de C. difficile que hiperproducen toxinas son responsables de un mayor nivel de morbilidad y mortalidad, ya que estas infecciones pueden ser resistentes a la terapia antimicrobiana y requerir colectomía. Debe considerarse la posibilidad de CDAD en todos los pacientes con diarrea asociada con el uso de antibióticos. Es necesario llevar un historial clínico detallado, ya que se ha notificado que la CDAD puede ocurrir hasta dos meses después de la administración de agentes antibacterianos.
En pacientes con disfunción renal grave (velocidad de filtración glomerular <10 ml/min), se observó un aumento del 33 % en la exposición sistémica a azitromicina.
La prolongación de la repolarización cardíaca y del intervalo QT, que aumenta el riesgo de arritmias cardíacas y de taquicardia ventricular/torsade de pointes (torsade de pointes), se ha observado durante el tratamiento con otros antibióticos macrólidos, incluyendo la azitromicina. Dado que las condiciones que cursan con mayor riesgo de arritmias ventriculares (incluyendo torsade de pointes) pueden provocar paro cardíaco, la azitromicina debe administrarse con precaución a pacientes con condiciones proarritmogénicas preexistentes (especialmente mujeres y pacientes de edad avanzada), en particular a pacientes:
- con prolongación congénita o registrada del intervalo QT;
- que actualmente estén recibiendo tratamiento con otras sustancias activas conocidas por prolongar el intervalo QT, por ejemplo antiarrítmicos de clase IA (quinidina y procainamida) y clase III (dofetilida, amiodarona y sotalol), cisaprida y terfenadina, neurolépticos como el pimozide; antidepresivos como el citalopram, y fluorquinolonas como la moxifloxacina y levofloxacina;
- con alteraciones del equilibrio electrolítico, especialmente en caso de hipokalemia e hipomagnesemia;
- con bradicardia clínicamente relevante, arritmia cardíaca o insuficiencia cardíaca grave.
Se han notificado empeoramiento de los síntomas de miastenia grave o desarrollo nuevo de síndrome miasténico en pacientes que reciben tratamiento con azitromicina.
En el tratamiento de faringitis/amonígelitis provocadas por Streptococcus pyogenes, el fármaco de elección generalmente es la penicilina, que también se utiliza para la prevención de la fiebre reumática aguda. La azitromicina es generalmente eficaz en el tratamiento de la infección estreptocócica de la orofaringe, pero no existen datos que demuestren la eficacia de la azitromicina en la prevención de los episodios reumáticos.
No se ha establecido la seguridad y eficacia de la administración intravenosa de azitromicina para el tratamiento de infecciones en niños.
No se ha establecido la seguridad y eficacia para la prevención o tratamiento de Mycobacterium Avium Complex en niños.
La azitromicina en forma de polvo para suspensión oral contiene sacarosa. No debe administrarse este medicamento a pacientes con trastornos hereditarios raros relacionados con la intolerancia a la fructosa, malabsorción de glucosa-galactosa o deficiencia de sacarasa-isomaltasa.
La azitromicina en forma de polvo para suspensión oral contiene 83,7 mg/dosis de fosfato de sodio. Debe tenerse precaución al administrarla a pacientes que siguen una dieta controlada en sodio.
Uso durante el embarazo o la lactancia.
Embarazo
No existen datos adecuados sobre el uso de azitromicina en mujeres embarazadas. En estudios de toxicidad reproductiva en animales no se observó efecto teratogénico adverso de la azitromicina sobre el feto, aunque el fármaco atraviesa la placenta. No se ha confirmado la seguridad del uso de azitromicina durante el embarazo. Por lo tanto, la azitromicina debe administrarse durante el embarazo solo si los beneficios superan los riesgos.
Lactancia
Se ha notificado que la azitromicina atraviesa la leche materna humana, pero no se han realizado estudios clínicos adecuados y bien controlados que permitan caracterizar la farmacocinética de la excreción de azitromicina en la leche materna humana.
Fertilidad
Se realizaron estudios de fertilidad en ratas; la tasa de embarazo disminuyó tras la administración de azitromicina. La relevancia de estos datos en humanos es desconocida.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
No hay evidencia de que la azitromicina pueda deteriorar la capacidad para conducir vehículos o manejar maquinaria, pero debe tenerse en cuenta la posibilidad de reacciones adversas como delirio, alucinaciones, vértigo, somnolencia, pérdida de conciencia o convulsiones, que podrían afectar dicha capacidad.
Vía de administración y dosis.
Sumamed® Forte, polvo para suspensión oral, se administra 1 vez al día, al menos 1 hora antes o 2 horas después de las comidas.
En caso de olvidar tomar una dosis, se debe tomar tan pronto como sea posible, y las siguientes dosis se deben tomar con intervalos de 24 horas.
Medición de la dosis
El envase contiene una jeringa graduada para dosificación oral. El volumen de la jeringa es de 5 ml.
Niños
En infecciones de los órganos ORL, del tracto respiratorio y de la piel y tejidos blandos (excepto eritema migrans crónico), la dosis diaria de azitromicina es de 10 mg/kg de peso corporal, lo que equivale a 0,25 ml/kg de peso corporal de la suspensión preparada. La duración del tratamiento es de 3 días.
Según el peso corporal del niño, se recomienda el siguiente esquema de dosificación de la suspensión
Sumamed® Forte:
| Masa corporal (kg) |
Dosis diaria de suspensión (ml) |
Frecuencia de administración |
Contenido de azitromicina en la dosis diaria de suspensión |
| 15–24 |
5 |
1 vez/día |
200 mg |
| 25–34 |
7,5 |
300 mg |
|
| 35–44 |
10 |
400 mg |
|
| ≥45 |
12,5 |
500 mg |
Preparación y uso de la suspensión
Al frasco que contiene el polvo, se debe añadir agua destilada o agua hervida y enfriada.
- Presione la tapa del frasco hacia abajo y gírela en sentido antihorario.
- Con una jeringa dosificadora, tome del recipiente limpio la cantidad adecuada de agua (9,5, 16,5 ó 20,0 ml) y añádala al frasco con el polvo:
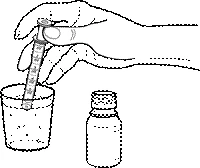
Frasco con polvo para suspensión oral 200 mg/5 ml de 15 ml: añadir 9,5 ml de agua al contenido del frasco. Agitar bien el contenido del frasco hasta obtener una suspensión homogénea. El volumen de la suspensión obtenida será aproximadamente 20 ml*.
Frasco con polvo para suspensión oral 200 mg/5 ml de 30 ml: añadir 16,5 ml de agua al contenido del frasco. Agitar bien el contenido del frasco hasta obtener una suspensión homogénea. El volumen de la suspensión obtenida será aproximadamente 35 ml*.
Frasco con polvo para suspensión oral 200 mg/5 ml de 37,5 ml: añadir 20 ml de agua al contenido del frasco. Agitar bien el contenido del frasco hasta obtener una suspensión homogénea. El volumen de la suspensión obtenida será aproximadamente 42,5 ml*.
* Tras disolver el polvo, el frasco contendrá 5 ml adicionales de suspensión (para compensar posibles pérdidas durante la administración).
| Volumen del frasco |
Cantidad de agua que debe añadirse al frasco para obtener la suspensión (ml) |
| 15 ml |
9,5 |
| 30 ml |
16,5 |
| 37,5 ml |
20 |
**La información sobre el volumen del frasco se encuentra en la caja y en la etiqueta del frasco.
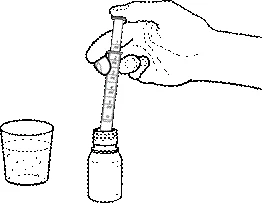
- Introducir la jeringa en la suspensión y, tirando del émbolo hacia arriba, medir la cantidad necesaria de suspensión.
- Si hay burbujas de aire en la jeringa, devolver el medicamento al frasco y repetir el procedimiento 3.
- Colocar al niño como para la alimentación.
- Colocar la punta de la jeringa en la boca del niño y expulsar lentamente el contenido.
- Permitir que el niño trague progresivamente toda la cantidad administrada.
|
|
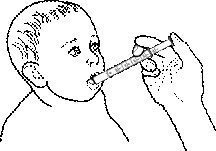
- Después de administrar el medicamento, darle al niño un poco de té o jugo para enjuagar y tragar los restos de la suspensión en la cavidad oral.
- Desmontar la jeringa utilizada, lavarla con agua corriente, secarla y guardarla en un lugar seco y limpio junto con el medicamento.
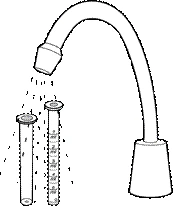
- Después de que el niño haya tomado la última dosis del medicamento, la jeringa y el frasco deben desecharse.
En caso de eritema migrans, la duración del tratamiento es de 5 días. El primer día debe administrarse 20 mg/kg de peso corporal de azitromicina, lo que equivale a 0,5 ml/kg de la suspensión preparada. Del día 2 al día 5, administrar 10 mg/kg de peso corporal, lo que equivale a 0,25 ml/kg de la suspensión preparada.
La dosis total del curso es de 60 mg/kg.
| Días de tratamiento |
1 |
2 |
3 |
4 |
5 |
| Dosis diaria (ml/kg) |
0,5 |
0,25 |
0,25 |
0,25 |
0,25 |
Se ha demostrado que la azitromicina es eficaz en el tratamiento de la faringitis estreptocócica en niños en forma de una dosis única de 10 mg/kg o 20 mg/kg durante 3 días. En estudios clínicos comparativos de estas dos dosis, se observó una eficacia clínica similar, aunque la erradicación bacteriana fue mayor con la dosis diaria de 20 mg/kg. Sin embargo, habitualmente el fármaco de elección para la profilaxis de la faringitis causada por Streptococcus pyogenes y de la poliartritis reumática que aparece como enfermedad secundaria es la penicilina.
Adultos
En infecciones de las vías respiratorias altas, tracto respiratorio inferior, piel y tejidos blandos (excepto eritema migrans crónico), la dosis total de azitromicina es de 1500 mg: 500 mg una vez al día. La duración del tratamiento es de 3 días.
En el eritema migrans, la dosis total de azitromicina es de 3 g: el primer día se debe tomar 1 g, seguido de 500 mg una vez al día del segundo al quinto día. La duración del tratamiento es de 5 días.
Pacientes de edad avanzada
No se requiere ajuste posológico en pacientes de edad avanzada.
Dado que los pacientes de edad avanzada pueden pertenecer a grupos de riesgo respecto a alteraciones de la conducción cardíaca, se recomienda precaución en el uso de azitromicina debido al riesgo de desarrollar arritmias cardíacas y arritmia tipo torsade de pointes.
Pacientes con alteración de la función renal
En pacientes con alteración leve de la función renal (velocidad de filtración glomerular de 10-80 ml/min) puede utilizarse la misma dosificación que en pacientes con función renal normal. La azitromicina debe administrarse con precaución en pacientes con alteración grave de la función renal (velocidad de filtración glomerular < 10 ml/min).
Pacientes con alteración de la función hepática
Dado que la azitromicina se metaboliza en el hígado y se elimina por la bilis, no debe administrarse a pacientes con alteración grave de la función hepática. No se han realizado estudios sobre el tratamiento con azitromicina en estos pacientes.
Niños.
Se recomienda administrar Sumamed® (100 mg/5 ml) a niños con peso corporal inferior a 15 kg. Sumamed® Forte se administra a niños con peso corporal superior a 15 kg.
Sobredosis.
La experiencia clínica con el uso de azitromicina indica que los efectos adversos que ocurren tras la ingesta de dosis superiores a las recomendadas son similares a los observados con las dosis terapéuticas habituales. Estos pueden incluir diarrea, náuseas, vómitos y pérdida reversible de la audición. En caso de sobredosis, si es necesario, se recomienda la administración de carbón activado y la realización de medidas terapéuticas generales sintomáticas y de apoyo.
Reacciones adversas.
En la tabla siguiente, clasificadas por órganos y sistemas y por frecuencia de aparición, se indican las reacciones adversas identificadas en estudios clínicos y durante el período de vigilancia poscomercialización, observadas tras la administración de todas las formas farmacéuticas de azitromicina. Las reacciones adversas registradas durante el período de vigilancia poscomercialización se indican en cursiva. Las categorías de frecuencia se definieron según la siguiente escala: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100 hasta < 1/10); poco frecuentes (≥ 1/1000 hasta < 1/100); raras (≥ 1/10000 hasta < 1/1000); muy raras (< 1/10000); frecuencia desconocida (no puede determinarse a partir de los datos disponibles). Dentro de cada grupo por frecuencia, los eventos adversos se enumeran en orden decreciente de gravedad.
Reacciones adversas posiblemente o probablemente relacionadas con azitromicina, basadas en datos obtenidos durante estudios clínicos y durante el período de vigilancia poscomercialización
| Clasificación por sistemas y órganos |
Reacción adversa |
Frecuencia |
| Infecciones e infestaciones |
Candidiasis, infecciones vaginales, neumonía, infección fúngica, infección bacteriana, faringitis, gastroenteritis, alteración de la función respiratoria, rinitis, candidiasis oral |
No frecuente |
| Colitis pseudomembranosa |
Desconocido |
|
| Alteraciones del sistema sanguíneo y del sistema linfático |
Leucopenia, neutropenia, eosinofilia |
No frecuente |
| Trombocitopenia, anemia hemolítica |
Desconocido |
|
| Alteraciones del sistema inmunitario |
Edema angioneurótico, reacciones de hipersensibilidad |
No frecuente |
| Reacción anafiláctica |
Desconocido |
|
| Alteraciones del metabolismo y nutrición |
Anorexia |
No frecuente |
| Alteraciones del estado psíquico |
Nerviosismo, insomnio |
No frecuente |
| Agitación |
Raro |
|
| Agresividad, ansiedad, delirio, alucinaciones |
Desconocido |
|
| Alteraciones del sistema nervioso |
Cefalea |
Frecuente |
| Vertigo, somnolencia, disgeusia, parestesia |
No frecuente |
|
| Pérdida de conciencia, convulsiones, hipoestesia, actividad psicomotora aumentada, anosmia, ageusia, parosmia, miastenia grave |
Desconocido |
|
| Alteraciones oculares |
Alteración de la visión |
No frecuente |
| Alteraciones del oído y del laberinto |
Alteraciones del oído, vértigo |
No frecuente |
| Alteración de la audición, incluyendo sordera y/o tinnitus |
Desconocido |
|
| Alteraciones cardíacas |
Palpitaciones |
No frecuente |
| Flutter /taquicardia ventricular torsade de pointes, arritmia, incluyendo taquicardia ventricular, prolongación del intervalo QT en el ECG |
Desconocido |
|
| Alteraciones vasculares |
Arores |
No frecuente |
| Hipotensión arterial |
Desconocido |
|
| Alteraciones del sistema respiratorio |
Disnea, epistaxis |
No frecuente |
| Alteraciones gastrointestinales |
Diarréa |
Muy frecuente |
| Vómitos, dolor abdominal, náuseas |
Frecuente |
|
| Estreñimiento, flatulencia, dispepsia, gastritis, disfagia, distensión abdominal, sequedad bucal, eructos, úlceras en la cavidad oral, hipersalivación |
No frecuente |
|
| Pancreatitis, cambio del color de la lengua |
Desconocido |
|
| Alteraciones del sistema hepato-biliar |
Alteración de la función hepática, ictericia colestásica |
Raro |
| Insuficiencia hepática (que rara vez ha conducido a desenlace letal), hepatitis fulminante, necrosis hepática |
Desconocido |
|
| Alteraciones de la piel y del tejido subcutáneo |
Erupción cutánea, prurito, urticaria, dermatitis, sequedad de la piel, hiperhidrosis |
No frecuente |
| Fotosensibilidad, erupción pustulosa exantemática aguda generalizada |
Raro |
|
| Síndrome de Stevens-Johnson, necrólisis epidérmica tóxica, eritema multiforme, reacción a medicamento con eosinofilia y síntomas sistémicos (DRESS) |
Desconocido |
|
| Alteraciones del sistema musculoesquelético |
Osteoartritis, mialgia, dolor de espalda, dolor de cuello |
No frecuente |
| Artralgia |
Desconocido |
|
| Alteraciones del sistema urinario |
Disuria, dolor renal |
No frecuente |
| Insuficiencia renal aguda, nefritis intersticial |
Desconocido |
|
| Alteraciones del sistema reproductor y de las glándulas mamarias |
Hemorragia uterina, alteraciones testiculares |
No frecuente |
| Alteraciones generales y alteraciones en el lugar de administración |
Edema, astenia, malestar general, fatiga, edema facial, dolor torácico, hipertermia, dolor, edema periférico |
No frecuente |
| Alteraciones en pruebas analíticas |
Disminución del número de linfocitos, aumento del número de eosinófilos, disminución del nivel de bicarbonato en sangre, aumento del nivel de basófilos, aumento del nivel de monocitos, aumento del nivel de neutrófilos |
Frecuente |
| Aumento del nivel de aspartato aminotransferasa, alanina aminotransferasa, bilirrubina en sangre, urea en sangre, creatinina en sangre; alteraciones en los niveles de potasio en sangre, aumento de la fosfatasa alcalina, cloruro, glucosa, plaquetas; disminución del nivel de hematocrito; aumento del nivel de bicarbonato, alteraciones en los niveles de sodio |
No frecuente |
|
| Lesiones e intoxicaciones |
Complicaciones tras el procedimiento |
No frecuente |
La información sobre reacciones adversas, posiblemente relacionadas con la profilaxis y el tratamiento del Mycobacterium Avium Complex, se basa en datos de estudios clínicos y en observaciones del período poscomercialización. Estas reacciones adversas difieren en tipo o frecuencia respecto a las informadas con la utilización de formas farmacéuticas de liberación inmediata y formas farmacéuticas de liberación prolongada.
Reacciones adversas, posiblemente relacionadas con la profilaxis y el tratamiento del Mycobacterium Avium Complex
| Clase de sistemas y órganos |
Reacción adversa |
Frecuencia |
||
| Trastornos del metabolismo |
Anorexia |
Frecuente |
||
| Trastornos del sistema nervioso |
Mareo, cefalea, parestesia, disgeusia |
Frecuente |
||
| Hipestesia |
Infrecuente |
|||
| Trastornos oculares |
Alteraciones visuales |
Frecuente |
||
| Trastornos del oído y del laberinto |
Sordera |
Frecuente |
||
| Alteraciones auditivas, tinnitus |
Infrecuente |
|||
| Trastornos cardiacos |
Palpitaciones |
Infrecuente |
||
| Trastornos gastrointestinales |
Diárea, dolor abdominal, náuseas, flatulencia, molestias gastrointestinales, evacuaciones frecuentes y líquidas |
Muy frecuente |
||
| Trastornos hepatobiliares |
Hepatitis |
Infrecuente |
||
| Trastornos de la piel y del tejido subcutáneo |
Erupción cutánea, prurito |
Frecuente |
||
| Síndrome de Stevens-Johnson, fotosensibilidad |
Infrecuente |
|||
| Trastornos del sistema musculoesquelético |
Artralgia |
Frecuente |
||
| Trastornos generales y alteraciones en el lugar de administración |
Aumento de la fatiga |
Frecuente |
||
| Astenia, malestar |
Infrecuente |
|||
Plazo de caducidad. 2 años.
El plazo de caducidad de la suspensión preparada es de 5 días (15 ml), 10 días (30 ml, 37,5 ml).
Condiciones de conservación.
Conservar a una temperatura no superior a 25 °C, en un lugar inaccesible para los niños.
Conservar la suspensión preparada a una temperatura no superior a 25 °C.
Envase.
Un frasco con polvo para suspensión oral de 15 ml (600 mg) o de 30 ml (1200 mg) o de 37,5 ml (1500 mg), junto con una jeringa dosificadora, en una caja.
Categoría de dispensación. Bajo receta médica.
Fabricante. PLIVA Hrvatska d.o.o.
Domicilio del fabricante y dirección del lugar de ejercicio de su actividad.
Prilaz baruna Filipovića 25, 10000 Zagreb, Croacia.