Sandostatin® LAR
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO SANDOSTATIN® LAR® (SANDOSTATIN® LAR®)
Composición:
Principio activo: octreótido en forma de acetato de octreótido;
1 frasco con polvo de 10 mg contiene 11,2 mg de acetato de octreótido, equivalente a 10 mg de octreótido;
1 frasco con polvo de 20 mg contiene 22,4 mg de acetato de octreótido, equivalente a 20 mg de octreótido;
1 frasco con polvo de 30 mg contiene 33,6 mg de acetato de octreótido, equivalente a 30 mg de octreótido;
Excipientes: poli (DL-láctido-co-glicólido), manitol (E 421);
disolvente para la preparación de la suspensión inyectable:
1 jeringa precargada de 2,0 ml contiene carmelosa sódica (carboximetilcelulosa sódica), manitol (E 421), agua para inyección, poloxámero 188.
Forma farmacéutica. Polvo para suspensión inyectable en conjunto con el disolvente.
Propiedades físico-químicas principales. Polvo de blanco a blanco con tono amarillento. Disolvente: solución de transparente a no más opalescente que la suspensión patrón I de la Farmacopea Europea, de color incoloro a ligeramente amarillo o marrón.
Grupo farmacoterapéutico. Preparados hormonales para uso sistémico (excepto hormonas sexuales e insulina). Hormonas hipofisarias, hipotalámicas y sus análogos. Hormonas hipotalámicas. Somatostatina y análogos. Octreótido.
Código ATC H01C B02.
Propiedades farmacológicas.
Farmacodinámica.
Octreótido es un octapéptido sintético, derivado del hormona natural somatostatina, con efectos farmacológicos similares, pero con una duración de acción significativamente más prolongada. El medicamento inhibe la secreción patológicamente elevada de la hormona del crecimiento (GH), así como de péptidos y serotonina producidos por el sistema endocrino gastroenteropancreático.
Los estudios en animales mostraron que el octreótido es un inhibidor más potente de la secreción de GH, glucagón e insulina que la somatostatina; el fármaco inhibe la secreción de GH y glucagón con mayor selectividad.
En voluntarios sanos, el octreótido, de forma similar a la somatostatina, inhibe:
- la secreción de GH estimulada por arginina, ejercicio físico e hipoglucemia inducida por insulina;
- la secreción de insulina, glucagón, gastrina y otros péptidos del sistema endocrino gastroenteropancreático estimulada por la ingesta de alimentos, así como la secreción de insulina y glucagón estimulada por arginina;
- la secreción de la hormona estimulante del tiroides (TSH), estimulada por el factor liberador de tirotropina (TRH).
A diferencia de la somatostatina, el octreótido inhibe la secreción de GH más que la de insulina, y su administración no se acompaña de una hipersecreción rebote de hormonas (por ejemplo, GH en pacientes con acromegalia).
En pacientes con acromegalia, Sandostatin® LAR, una formulación farmacéutica de liberación prolongada de octreótido, puede administrarse con intervalos de 4 semanas, lo que proporciona niveles terapéuticos constantes de octreótido en suero, reduciendo progresivamente los niveles de GH y normalizando la concentración del factor de crecimiento similar a la insulina-1 (IGF-1) en suero en la mayoría de los pacientes. En la mayoría de los pacientes, Sandostatin® LAR reduce notablemente las manifestaciones clínicas de la enfermedad, tales como cefalea, sudoración excesiva, parestesias, fatiga, osteoartalgia y síndrome del túnel carpiano. En una proporción considerable (50 %) de pacientes con acromegalia no tratados previamente y con adenoma hipofisario con hipersecreción de GH, el tratamiento con Sandostatin® LAR condujo a una reducción del volumen tumoral > 20 %. Además, estudios a corto plazo indican que el uso de octreótido antes del tratamiento quirúrgico en algunos pacientes con adenoma hipofisario puede conducir a una reducción del tamaño tumoral. Sin embargo, el tratamiento quirúrgico no debe retrasarse.
En pacientes con tumores endocrinos activos del tracto gastrointestinal y del páncreas, el tratamiento con Sandostatin® LAR proporciona un control sostenido de varios síntomas clínicos relacionados con la enfermedad. A continuación se describe el efecto del octreótido sobre diferentes tipos de tumores endocrinos del tracto gastrointestinal y del páncreas.
Tumores carcinoides.
La administración de octreótido puede aliviar los síntomas, especialmente los sofocos y la diarrea. En muchos casos, esto se asocia con una disminución de la concentración de serotonina en plasma y una reducción en la excreción urinaria de ácido 5-hidroxindolacético.
Tumores caracterizados por la hiperproducción del péptido intestinal vasoactivo (VIP).
La característica bioquímica de estos tumores es la producción excesiva de VIP. En la mayoría de los casos, el uso de octreótido reduce la gravedad de la diarrea secretora, típica de esta patología, mejorando así la calidad de vida del paciente. Esto se asocia con una reducción de las alteraciones electrolíticas asociadas, como la hipokalemia, lo que permite suspender la administración oral y parenteral de líquidos y electrolitos. En algunos pacientes, según tomografía computarizada, se observa una desaceleración o detención de la progresión tumoral e incluso una reducción del tamaño, especialmente de las metástasis hepáticas. La mejoría clínica suele acompañarse de una disminución (hasta valores normales) de la concentración de VIP en plasma.
Glucagonomas.
La administración de octreótido conduce, en la mayoría de los casos, a una reducción significativa de las erupciones migratorias necrolíticas, características de esta enfermedad. El octreótido no tiene un efecto notable sobre la gravedad de la diabetes mellitus, que con frecuencia se observa en los glucagonomas, y generalmente no reduce la necesidad de insulina o de fármacos hipoglucemiantes orales. En pacientes con diarrea, el octreótido reduce su gravedad, lo que se asocia con un aumento del peso corporal.
Con el uso de octreótido, frecuentemente se observa una disminución rápida de la concentración de glucagón en plasma, aunque este efecto no se mantiene con el tratamiento prolongado. No obstante, la mejoría sintomática permanece estable durante un período prolongado.
Gastrinomas / síndrome de Zollinger-Ellison.
Principalmente, la terapia con inhibidores de la bomba de protones o bloqueadores de los receptores de histamina H2 inhibe la hipersecreción de ácido clorhídrico en el estómago. Sin embargo, los inhibidores de la bomba de protones o los bloqueadores de los receptores H2 de histamina no siempre logran aliviar adecuadamente la diarrea, que también es un síntoma importante de la enfermedad. En algunos pacientes, Sandostatin® LAR contribuye a una mayor reducción de la hipersecreción de ácido clorhídrico y a una mejoría clínica, incluida la diarrea, mediante la disminución de la concentración elevada de gastrina.
Insulinomas.
La administración de octreótido reduce el nivel de insulina inmunorreactiva en sangre. En pacientes con tumores operables, el octreótido puede ayudar a restaurar y mantener la normoglucemia en el período preoperatorio. En pacientes con tumores benignos o malignos no operables, el control glucémico puede mejorar sin necesidad de una reducción sostenida del nivel de insulina en sangre.
Tumores neuroendocrinos metastásicos del intestino o tumores primarios de localización desconocida, cuando se han excluido otras localizaciones primarias distintas del intestino.
Un estudio aleatorizado, doble ciego, controlado con placebo de fase III (PROMID) mostró que Sandostatin® LAR inhibe el crecimiento tumoral en pacientes con tumores neuroendocrinos metastásicos del intestino medio (intestino delgado, apéndice, ciego y porción ascendente del colon). 85 pacientes fueron aleatorizados a recibir Sandostatin® LAR 30 mg cada 4 semanas (n = 42) o placebo (n = 43) durante 18 meses o hasta la progresión tumoral o la muerte.
Los criterios principales de inclusión fueron: ausencia de tratamiento previo, diagnóstico histológicamente confirmado, tumores bien diferenciados localmente no operables o metastásicos; tumores neuroendocrinos/carcinomas funcionales o no funcionales; localización del tumor primario en el intestino medio (intestino delgado, apéndice, ciego y porción ascendente del colon) o tumor primario de localización desconocida atribuido al intestino medio, cuando se excluyeron localizaciones primarias en el páncreas, tórax u otros sitios.
El punto final primario fue el tiempo hasta la progresión tumoral o la muerte por causa tumoral (TTP).
El análisis por intención de tratar (ITT) de todos los pacientes aleatorizados mostró que 26 eventos de progresión o muertes relacionadas con el tumor ocurrieron en el grupo Sandostatin® LAR, mientras que 41 ocurrieron en el grupo placebo (HR = 0,32; IC 95 %: 0,19 – 0,55; p = 0,000015).
En un análisis conservador ITT, excluyendo 3 pacientes desviados del aleatorización, se observaron 26 y 40 eventos de progresión o muerte por causa tumoral en los grupos Sandostatin® LAR y placebo, respectivamente (HR = 0,34; IC 95 %: 0,20–0,59; valor p = 0,000072; figura).
En la población del análisis conservador ITT (cITT), en la que 3 pacientes fueron excluidos durante la aleatorización, se observaron 26 y 40 eventos de progresión o muerte por causa tumoral en los grupos Sandostatin® LAR y placebo, respectivamente.
La mediana del tiempo hasta la progresión tumoral fue de 14,3 meses (IC 95 %: 11,0 – 28,8 meses) en el grupo Sandostatin® LAR y de 6 meses (IC 95 %: 3,7 – 9,4 meses) en el grupo placebo.
En el análisis de la población según protocolo, en el que se excluyeron pacientes adicionales al final del tratamiento recibido en el estudio, se observaron 19 y 38 eventos de progresión tumoral o muerte por causa tumoral en los grupos Sandostatin® LAR y placebo, respectivamente (HR = 0,24; IC 95 %: 0,13 – 0,45; valor p = 0,0000036).
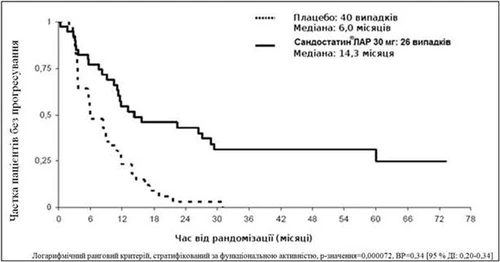
Figura. Estimación del tiempo hasta la progresión tumoral según el método de Kaplan-Meier en los grupos Sandostatin® LAR y placebo (población del análisis conservador ITT)
Tabla 1
Indicadores del tiempo hasta el inicio de la progresión tumoral según el análisis poblacional
| Análisis |
Casos de TTP |
TTP medio, meses [IC del 95 %] |
RR [IC del 95 %], p* |
||
| Sandostatin® LAR |
Placebo |
Sandostatin® LAR |
Placebo |
||
| ITT |
26 |
41 |
ND |
ND |
0,32 [IC del 95 %: 0,19-0,55] p = 0,000015 |
| cITT |
26 |
40 |
14,3 [IC del 95 %: 11,0 – 28,8] |
6,0 [IC del 95 %: 3,7-9,4] |
0,34 [IC del 95 %: 0,20-0,59] p = 0,000072 |
| ND – no disponible; RR – riesgo relativo; TTP – tiempo hasta la progresión tumoral; ITT – análisis por intención de tratar; cITT – análisis conservador por intención de tratar; *prueba de rango logarítmico, estratificada por actividad funcional |
|||||
El efecto del tratamiento fue similar en pacientes con tumores funcionalmente activos (HR=0,23; IC del 95 %: 0,09 – 0,57) y en aquellos con tumores inactivos (HR=0,25; IC del 95 %: 0,10 – 0,59). Tras 6 meses de tratamiento, la estabilidad de la enfermedad se observó en el 66 % de los pacientes del grupo de Sandostatina® LAR y en el 37 % de los pacientes del grupo placebo.
Debido a la eficacia clínica significativa demostrada para Sandostatina® LAR en este análisis intermedio previamente planificado, se interrumpió el reclutamiento de participantes en el estudio.
La seguridad de Sandostatina® LAR en este estudio fue coherente con su perfil de seguridad establecido.
Farmacocinética.
Tras una inyección intramuscular única de Sandostatina® LAR, se alcanza temporalmente un pico inicial de la concentración de octreótido en suero dentro de la primera hora tras la administración, seguido de una disminución en las siguientes 24 horas hasta niveles no detectables. Tras este pico inicial del día 1, en la mayoría de los pacientes la concentración de octreótido permanece en niveles subterapéuticos durante los siguientes 7 días. Posteriormente, la concentración de octreótido aumenta nuevamente, alcanzando una meseta aproximadamente el día 14 y manteniéndose en este nivel durante las siguientes 3 – 4 semanas. La concentración pico del día 1 es inferior a las concentraciones durante la fase de meseta y no supera el 0,5 % de la liberación total del fármaco en el día 1. Aproximadamente al día 42, la concentración de octreótido disminuye lentamente, coincidiendo con la fase terminal de degradación de la matriz polimérica de la forma farmacéutica.
En pacientes con acromegalia, la concentración de octreótido en la fase de meseta tras una administración única de Sandostatina® LAR en dosis de 10, 20 o 30 mg fue de 358, 926 y 1710 ng/l, respectivamente. La concentración de octreótido en suero en estado estacionario, alcanzada tras 3 inyecciones con intervalos de 4 semanas, es 1,6 a 1,8 veces mayor y alcanza hasta 1557 ng/l y 2384 ng/l tras administraciones múltiples de 20 y 30 mg de Sandostatina® LAR, respectivamente.
En pacientes con tumores carcinoides, la media aritmética (y la mediana) de la concentración de octreótido en suero en estado estacionario tras las dosis de 10, 20 y 30 mg de Sandostatina® LAR fue de 1231 (894), 2620 (2270) y 3928 (3010) ng/l, respectivamente.
No se observó acumulación de octreótido, tal como se esperaba por la superposición de los perfiles de liberación tras inyecciones mensuales de Sandostatina® LAR durante 28 meses.
El perfil farmacocinético del octreótido tras la inyección de Sandostatina® LAR refleja el perfil de liberación desde la matriz polimérica y su degradación. Inmediatamente tras su liberación en la circulación sistémica, el octreótido se distribuye según sus propiedades farmacocinéticas descritas para la administración subcutánea. El volumen de distribución del octreótido en estado estacionario es de 0,27 l/kg, y el aclaramiento total es de 160 ml/min. La unión a las proteínas plasmáticas alcanza el 65 %, mientras que la unión a los elementos formes de la sangre es muy baja.
Características clínicas.
Indicaciones.
Acromegalia (en pacientes con experiencia previa en el uso del medicamento Sandostatin®):
- tratamiento de las manifestaciones principales de la enfermedad cuando no se ha logrado un efecto suficiente mediante cirugía o radioterapia;
- tratamiento entre ciclos de radioterapia hasta que se alcance su efectividad.
Síntomas de tumores endocrinos funcionales del tracto gastrointestinal (en pacientes con experiencia previa en el uso del medicamento Sandostatin®):
- tumores carcinoides con síndrome carcinoide presente;
- VIPomas;
- glucagonomas;
- gastrinomas/síndrome de Zollinger-Ellison;
- insulinomas, para el control de la hipoglucemia en el período preoperatorio, así como para terapia de mantenimiento;
- somatolibreinomas (tumores que producen factor liberador de hormona del crecimiento).
Tumores neuroendocrinos metastásicos del intestino o con localización primaria desconocida, cuando se han excluido otras localizaciones primarias distintas del intestino.
Contraindicaciones.
Hipersensibilidad individual al octreótido o a cualquiera de los excipientes del medicamento (incluyendo el disolvente).
Interacción con otros medicamentos y otras formas de interacción.
Puede ser necesaria la ajuste de la dosis de medicamentos tales como betabloqueantes, bloqueantes de los canales de calcio o agentes para el control del equilibrio hídrico y electrolítico durante el tratamiento concomitante con Sandostatin® LAR.
Puede ser necesaria la ajuste de la dosis de insulina y medicamentos hipoglucemiantes durante el tratamiento concomitante con Sandostatin® LAR.
Se ha demostrado que el octreótido reduce la absorción intestinal de ciclosporina y retrasa la absorción de cimetidina.
La administración concomitante de octreótido y bromocriptina aumenta la biodisponibilidad de la bromocriptina.
Datos publicados limitados indican que los análogos de la somatostatina pueden reducir el aclaramiento metabólico de sustancias metabolizadas por las enzimas del citocromo P450, lo cual podría deberse a la supresión de la secreción de GH. Dado que no puede descartarse este efecto del octreótido, se debe tener precaución al administrar otros medicamentos que se metabolizan principalmente por CYP3A4, así como aquellos con un índice terapéutico estrecho (por ejemplo, quinidina, terfenadina).
Características de uso.
Generales
Dado que, en ocasiones, el tamaño de los tumores hipofisarios productores de GH puede aumentar, provocando complicaciones graves (por ejemplo, estrechamiento del campo visual), es fundamental realizar un seguimiento cuidadoso de todos los pacientes. En caso de presentarse signos de aumento del tamaño del tumor, se debe considerar la necesidad de aplicar tratamientos alternativos.
El efecto terapéutico de la reducción de los niveles de GH y la normalización de la concentración de IGF-1 en mujeres con acromegalia podría potencialmente restaurar la fertilidad. Durante el tratamiento con octreótido, se debe recomendar a las mujeres en edad reproductiva el uso de métodos anticonceptivos adecuados si fuera necesario.
En pacientes que reciben tratamiento prolongado con octreótido, se debe controlar la función tiroidea.
Durante la terapia con octreótido, se debe monitorear la función hepática.
Eventos relacionados con el sistema cardiovascular
Se han notificado frecuentemente casos de bradicardia. Puede ser necesario ajustar la dosis de fármacos como betabloqueadores, bloqueadores de los canales de calcio, o medicamentos que regulan el equilibrio de líquidos o electrolitos.
Se han observado casos de bloqueo auriculoventricular (incluyendo bloqueo auriculoventricular completo) en pacientes que recibieron infusiones continuas de dosis altas (100 microgramos/hora) y en pacientes que recibieron octreótido por vía intravenosa en forma de bolo (50 microgramos como bolo, seguido de 50 microgramos/hora por infusión continua). Por lo tanto, no se debe exceder la dosis máxima de 50 microgramos/hora (ver Sección «Vía de administración y dosis»). Los pacientes que reciban octreótido por vía intravenosa en dosis altas deben estar bajo monitoreo cardíaco adecuado.
Eventos relacionados con la vesícula biliar
La litiasis biliar es la complicación más frecuente del tratamiento con Sandostatin® y puede estar asociada con colecistitis y engrosamiento de la pared de la vesícula biliar (ver sección «Reacciones adversas»). Además, se han notificado casos de colangitis como complicación de la litiasis biliar en pacientes que recibieron Sandostatin® LAR durante el período poscomercialización. Se recomienda realizar una ecografía de la vesícula biliar antes del inicio del tratamiento con Sandostatin® LAR y aproximadamente cada 6 meses durante el tratamiento.
Función del páncreas
Se ha observado insuficiencia pancreática exocrina (IPE) en algunos pacientes que recibieron tratamiento con octreótido para tumores neuroendocrinos gastroenteropancreáticos. Los síntomas de IPE pueden incluir esteatorrea, diarrea, distensión abdominal y pérdida de peso. Se debe considerar la realización de pruebas de cribado y el tratamiento adecuado de la IPE según las recomendaciones clínicas en pacientes con síntomas.
Metabolismo de la glucosa
Debido a la supresión de los niveles de GH y la liberación de glucagón e insulina, Sandostatin® LAR puede alterar la regulación de la glucosa. Puede alterarse la tolerancia a la glucosa tras la ingesta de alimentos. En algunos casos, con la administración prolongada, puede desarrollarse hiperglucemia persistente, como se ha observado en pacientes que recibieron Sandostatin® por vía subcutánea. También puede ocurrir hipoglucemia.
En pacientes con diabetes mellitus tipo I concomitante, Sandostatin® LAR puede afectar el control glucémico, reduciendo la necesidad de insulina. En pacientes sin diabetes y en pacientes con diabetes mellitus tipo II con reserva de insulina parcialmente alterada, la administración subcutánea de Sandostatin® puede provocar un empeoramiento de la glucemia tras la ingesta de alimentos. Por ello, se recomienda el monitoreo de la tolerancia a la glucosa y el tratamiento antidiabético.
Dado que el octreótido tiene, en comparación con la insulina, un efecto supresor relativamente mayor sobre la GH y el glucagón, y una duración de acción inhibitoria más corta sobre la secreción de insulina, en pacientes con insulinaoma, el octreótido puede aumentar la profundidad y duración de la hipoglucemia. El estado de estos pacientes debe vigilarse cuidadosamente.
Alimentación
El octreótido puede alterar en algunos pacientes la absorción de las grasas dietéticas.
En algunos pacientes que reciben tratamiento con octreótido, se han observado niveles reducidos de vitamina B12 y resultados anormales en la prueba de Schilling. En pacientes con antecedentes de deficiencia de vitamina B12, se debe controlar el nivel de esta vitamina durante el tratamiento con Sandostatin® LAR.
Contenido de sodio
Sandostatin® LAR contiene menos de 1 mmol (23 mg) de sodio por dosis, por lo que puede considerarse un medicamento sin sodio.
Uso durante el embarazo o la lactancia.
Los datos sobre el uso de octreótido en mujeres embarazadas son limitados (menos de 300 embarazos), y aproximadamente en un tercio de los casos los resultados del embarazo son desconocidos. La mayoría de los informes provienen del uso poscomercialización de octreótido, y la proporción de mujeres embarazadas entre las pacientes con acromegalia fue superior al 50%. La mayoría de las mujeres recibieron octreótido durante el primer trimestre del embarazo en dosis de 100 a 1200 microgramos/día en forma de Sandostatin® subcutáneo o de 10 a 40 mg/mes de Sandostatin® LAR. Las malformaciones congénitas se registraron en aproximadamente el 4% de los embarazos, según los casos con resultados conocidos. En estos casos, no se ha sospechado una relación causal con el octreótido.
En estudios en animales no se han observado efectos tóxicos reproductivos directos ni indirectos.
Como medida de precaución, se recomienda evitar el uso de Sandostatin® durante el embarazo.
Lactancia
No se sabe si el octreótido pasa a la leche materna humana. Estudios en animales han demostrado la excreción de octreótido en la leche materna. Las pacientes que reciben tratamiento con Sandostatin® LAR deben suspender la lactancia.
Fertilidad
No se conoce si el octreótido afecta la fertilidad humana. En recién nacidos masculinos de pacientes tratadas con el medicamento durante el embarazo y la lactancia, se observó retardo en el descenso de los testículos. Sin embargo, en estudios experimentales en ratas macho y hembra, el octreótido no afectó la fertilidad a dosis de hasta 1 mg/kg de peso corporal por día.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
Sandostatin® LAR no tiene o tiene un efecto insignificante sobre la capacidad para conducir vehículos o manejar maquinaria. No obstante, se debe recomendar a los pacientes tener precaución al conducir automóviles o trabajar con maquinaria si experimentan mareos, astenia/fatiga o cefalea durante el tratamiento con Sandostatin® LAR.
Vía de administración y dosis.
Acrómegalia
Se recomienda iniciar el tratamiento con Sandostatin® LAR con una dosis de 20 mg durante 3 meses, con intervalos entre inyecciones de 4 semanas. Los pacientes que están siendo tratados con Sandostatin® pueden comenzar el tratamiento con Sandostatin® LAR al día siguiente de la última administración subcutánea de Sandostatin®. El ajuste posterior de la dosis se realizará en función de los niveles plasmáticos de GH, la concentración de IGF-1 y los síntomas clínicos de la enfermedad.
En los pacientes en los que, durante este período de 3 meses, los síntomas clínicos y los parámetros bioquímicos (GH, IGF-1) no se controlen completamente (las concentraciones de GH permanecen por encima de 2,5 µg/l), la dosis puede aumentarse hasta 30 mg cada 4 semanas. Si tras un período de tratamiento de 3 meses con dosis de 30 mg las concentraciones de GH, IGF-1 y/o los síntomas no están completamente controlados, la dosis puede aumentarse hasta 40 mg cada 4 semanas.
En los pacientes en los que tras 3 meses de tratamiento con 20 mg del fármaco la concentración de GH se mantiene por debajo de 1 µg/l, se normaliza la concentración de IGF-1 en suero y desaparecen los síntomas previos de acromegalia, puede administrarse 10 mg de Sandostatin® LAR cada 4 semanas. Sin embargo, en estos pacientes que reciben una dosis baja de Sandostatin® LAR se recomienda intensificar el control de las concentraciones de GH e IGF-1, así como de los síntomas clínicos de la enfermedad.
La evaluación de las concentraciones de GH e IGF-1 en pacientes que reciben una dosis estable de Sandostatin® LAR debe realizarse cada 6 meses.
Tumores endocrinos del tracto gastrointestinal y del páncreas
Tratamiento de pacientes con síntomas relacionados con tumores neuroendocrinos funcionales del tracto gastrointestinal y del páncreas.
Se recomienda iniciar el tratamiento con una dosis de 20 mg de Sandostatin® LAR durante 3 meses, con intervalos entre inyecciones de 4 semanas. Tras la primera inyección de Sandostatin® LAR, debe continuarse durante 2 semanas la administración subcutánea de Sandostatin® en la dosis eficaz previamente utilizada.
En los pacientes en los que los síntomas y los marcadores biológicos estén adecuadamente controlados tras 3 meses de tratamiento, la dosis de Sandostatin® LAR puede reducirse a 10 mg cada 4 semanas.
En los pacientes en los que los síntomas y los marcadores biológicos solo estén parcialmente controlados tras 3 meses de tratamiento, la dosis puede aumentarse a 30 mg cada 4 semanas.
En los días en los que durante el tratamiento con Sandostatin® LAR empeoren los síntomas relacionados con tumores del tracto gastrointestinal y del páncreas, se recomienda la administración adicional de Sandostatin® por vía subcutánea en la dosis que se utilizaba antes del inicio del tratamiento con Sandostatin® LAR. Esto puede ocurrir principalmente durante los primeros 2 meses de tratamiento, hasta que se alcance la concentración terapéutica de octreótido.
Tratamiento de pacientes con tumores neuroendocrinos metastásicos del intestino medio o con tumor primario de localización desconocida, cuando se han excluido otras localizaciones primarias distintas del intestino medio.
La dosis recomendada de Sandostatin® LAR es de 30 mg una vez cada 4 semanas. El tratamiento con Sandostatin® LAR para el control de la enfermedad tumoral debe continuar incluso en ausencia de progresión de la enfermedad.
Uso en pacientes con insuficiencia renal
La alteración de la función renal no afecta a la exposición sistémica total al octreótido durante su administración en forma de Sandostatin® por vía subcutánea. Por tanto, no es necesaria la ajuste de la dosis de Sandostatin® LAR en estos pacientes.
Uso en pacientes con insuficiencia hepática
En estudios con administración subcutánea e intravenosa de Sandostatin®, se ha observado que en pacientes con cirrosis hepática la eliminación del fármaco puede estar reducida, lo cual no ocurre en pacientes con esteatosis hepática. En algunos casos, puede ser necesaria la ajuste de la dosis de Sandostatin® LAR en pacientes con alteración de la función hepática.
Uso en pacientes de edad avanzada
En estudios con administración subcutánea de Sandostatin® se ha observado que en pacientes de 65 años o más no es necesaria la ajuste de la dosis.
Vía de administración
Sandostatin® LAR está indicado para tratamiento prolongado según las indicaciones del médico.
Para minimizar al máximo el dolor en el lugar de inyección, se recomienda dejar que el fármaco Sandostatin® LAR alcance la temperatura ambiente antes de su administración.
Para la preparación de la suspensión de Sandostatin® LAR y la administración de una inyección intramuscular profunda en el músculo glúteo, deben utilizarse los componentes del kit de inyección.
Sandostatin® LAR se administra mediante inyección intramuscular profunda. En caso de administración repetida del fármaco, se recomienda alternar el lugar de inyección, rotando entre los músculos glúteos derecho e izquierdo.
Instrucciones para la administración de inyecciones intramusculares de Sandostatin® LAR
exclusivamente para inyección intramuscular profunda en el músculo glúteo:
Contenido del envase:
**
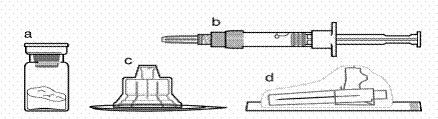
**
a un frasco que contiene el fármaco Sandostatin® LAR
b una jeringa precargada con disolvente
c un adaptador para frasco en un contenedor de plástico
d una aguja segura para inyecciones (0,91 mm × 38,1 mm; 19G × 1,5") con tapa protectora
| Siga cuidadosamente las instrucciones que se indican a continuación para la correcta preparación del medicamento Sandostatin® LAR antes de su administración por vía intramuscular profunda. La suspensión de Sandostatin® LAR debe prepararse inmediatamente antes de su uso. La inyección debe ser administrada únicamente por personal médico experimentado. |
|
|
|
Paso 1 Extraiga el medicamento Sandostatin® LAR del refrigerador y asegúrese de que alcance la temperatura ambiente. Habitualmente, esto puede tardar entre 30 y 60 minutos, pero no más de 24 horas. Lávese las manos con agua tibia y jabón. Coloque el kit sobre una superficie limpia y plana. Retire la película protectora de la bandeja que contiene el conjunto para inyección. Retire la tapa del frasco que contiene el medicamento Sandostatin® LAR. |
|
|
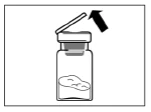
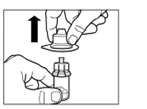
Paso 2 Limpie el tapón de goma del frasco con una torunda de alcohol. Advertencia: no toque el tapón de goma después de haberlo limpiado con alcohol. Retire la película protectora del recipiente plástico que contiene el adaptador para frasco. NO retire el adaptador del recipiente plástico. Coloque el recipiente plástico con el adaptador encima del frasco y presione hacia abajo hasta que escuche un clic característico. Retire verticalmente hacia arriba el recipiente plástico del adaptador para frasco. |
|
|
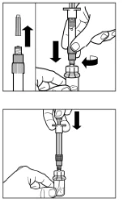
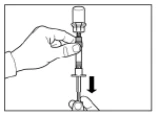
Paso 3 Retire la tapa de la jeringa que contiene el disolvente y enrosque la jeringa en el adaptador para frasco. Presione lentamente el émbolo hacia abajo hasta el fondo para transferir todo el disolvente al frasco. |
|
|
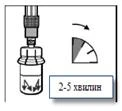
Paso 4 Deje el frasco en posición vertical hasta que el disolvente humedezca completamente el medicamento Sandostatin® LAR (no menos de 2 a 5 minutos). Durante este tiempo, prepare al paciente para la inyección. ¡Tenga en cuenta! Es normal que, debido a una ligera presión interna en el frasco, el émbolo de la jeringa suba ligeramente. |
|
|
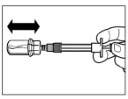
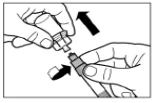
Paso 5 Después de la disolución, presione nuevamente el émbolo de la jeringa hacia abajo hasta el fondo. Manteniendo el émbolo presionado, agite suavemente el frasco en movimiento horizontal durante aproximadamente 30 segundos para mezclar su contenido. Verifique visualmente que el medicamento se haya disuelto completamente (debe formarse una suspensión homogénea de aspecto lechoso). Si no se ha disuelto completamente, continúe agitando el frasco durante otros 30 segundos. |
|
|
Paso 6 Coloque la jeringa y el frasco boca abajo, lentamente tire del émbolo hacia usted para transferir el contenido del frasco a la jeringa. Desenrosque la jeringa del adaptador para frasco. El medicamento debe administrarse inmediatamente después de su preparación. |
|
|
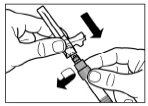
Paso 7 Enrosque la aguja en la jeringa. Retire la tapa protectora de la aguja tirando de ella. Gire cuidadosamente la jeringa, manteniendo la homogeneidad de su contenido. Dele ligeros golpes con el dedo sobre la jeringa para eliminar todas las burbujas visibles. La solución reconstituida de Sandostatin® LAR está lista para su uso inmediato. |
|
|
Paso 8 Sandostatin® LAR solo debe administrarse mediante inyección intramuscular profunda. Está contraindicada la administración intravenosa del medicamento. Limpie el sitio de inyección con una torunda de alcohol. Introduzca completamente la aguja en el glúteo derecho o izquierdo. Tire suavemente del émbolo de la jeringa hacia atrás para asegurarse de que la aguja no haya penetrado en un vaso sanguíneo; en caso contrario, cambie la posición de la aguja. Presione lentamente el émbolo de la jeringa para administrar la dosis requerida del medicamento. Tras finalizar la inyección, retire la aguja del sitio de inyección y active el mecanismo de seguridad como se muestra a continuación. |
|
|
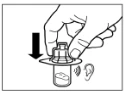
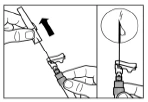
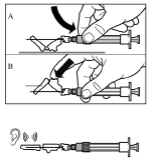
Paso 9 Active el mecanismo de seguridad de la aguja con una sola mano, utilizando uno de los métodos descritos a continuación: A. Presione la parte abatible del dispositivo de seguridad contra una superficie dura, por ejemplo, una mesa. B. Presione con el dedo índice la parte abatible del dispositivo de seguridad. Recuerde que, en todo momento, debe mantener los dedos alejados de la cánula de la aguja. El sonido de clic confirma la activación correcta del mecanismo de seguridad. Deseche inmediatamente el frasco, la jeringa y la aguja en un contenedor para objetos punzantes o en cualquier otro recipiente resistente y cerrado destinado al desecho. |
Niños.
La administración del medicamento Sandostatina® LAR está contraindicada en niños debido a la falta de experiencia clínica.
Sobredosis.
Se han notificado un número limitado de casos de sobredosis accidental con Sandostatina® LAR (en un rango de dosis de 100 – 163 mg/mes). El único efecto adverso observado fueron sofocos. También se han notificado pacientes con cáncer que recibieron dosis de Sandostatina® LAR hasta de 60 mg/mes y hasta de 90 mg/2 semanas. Estas dosis fueron generalmente bien toleradas. Sin embargo, se han registrado efectos adversos como poliuria, fatiga, depresión, ansiedad y dificultad de concentración.
El tratamiento es sintomático.
Reacciones adversas.
Caracterización breve del perfil de seguridad del medicamento
Las reacciones adversas más frecuentes durante el tratamiento con octreótido afectan al sistema gastrointestinal, sistema nervioso, hígado y vesícula biliar, así como al metabolismo y a la nutrición.
Las reacciones adversas más comunes observadas en los estudios clínicos con octreótido fueron diarrea, dolor abdominal, náuseas, flatulencia, cefalea, litiasis biliar, hiperglucemia y estreñimiento. Otras reacciones adversas frecuentes incluyeron mareo, dolor local, cálculos biliares, disfunción tiroidea (por ejemplo, niveles reducidos de TSH, niveles reducidos de T4 total y de T4 libre), deposiciones líquidas, alteraciones en la tolerancia a la glucosa, vómitos, astenia e hipoglucemia.
En ocasiones, los efectos adversos gastrointestinales pueden simular una obstrucción intestinal aguda con distensión abdominal progresiva, intenso dolor en la región epigástrica, sensibilidad y tensión muscular en la región abdominal.
Aunque la excreción fecal de grasa puede aumentar, no existen datos que indiquen que el tratamiento prolongado con Sandostatin® LAR pueda provocar deficiencias nutricionales debidas a alteraciones en la absorción (malabsorción).
Muy raramente se han notificado casos de pancreatitis aguda, que se desarrollaron durante las primeras horas o días de tratamiento con Sandostatin® LAR y que desaparecieron tras la interrupción del fármaco. Asimismo, se han notificado casos de pancreatitis provocada por litiasis biliar en pacientes sometidos a tratamiento prolongado con Sandostatin® LAR.
Tanto en pacientes con acromegalia como con síndrome carcinoide, durante la terapia con octreótido se han observado cambios en el ECG tales como prolongación del intervalo QT, desviaciones del eje, repolarización ventricular temprana, bajo voltaje, transición R/S, aumento precoz de la onda R y cambios inespecíficos en la onda ST-T. No se ha establecido una relación causal entre estos hallazgos y el octreótido, ya que muchos de estos pacientes presentan enfermedades cardiológicas subyacentes.
Las reacciones adversas que se indican a continuación se han registrado durante ensayos clínicos con octreótido y se presentan según su frecuencia: muy frecuentes (≥ 1/10); frecuentes (≥ 1/100, < 1/10); poco frecuentes (≥ 1/1000, < 1/100); raras (≥ 1/10000, < 1/1000); muy raras (< 1/10000), incluyendo informes aislados. Dentro de cada grupo de frecuencia, las reacciones adversas se enumeran en orden decreciente de gravedad.
Reacciones adversas al medicamento notificadas durante estudios clínicos
| Alteraciones del sistema gastrointestinal Muy frecuentes: diarrea, dolor abdominal, náuseas, estreñimiento, flatulencia. Frecuentes: dispepsia, vómitos, distensión abdominal, esteatorrea, evacuaciones frecuentes y líquidas, decoloración de las heces. |
| Alteraciones del sistema nervioso Muy frecuentes: cefalea. Frecuentes: vértigo. |
| Alteraciones del sistema endocrino Frecuentes: hipotiroidismo, alteración de la función tiroidea (por ejemplo, niveles reducidos de TSH, niveles reducidos de T4 total, niveles reducidos de T4 libre). |
| Alteraciones del sistema hepatobiliar Muy frecuentes: litiasis biliar. Frecuentes: colecistitis, cálculos biliares, hiperbilirrubinemia. |
| Alteraciones del metabolismo y de la nutrición Muy frecuentes: hiperglucemia. Frecuentes: hipoglucemia, alteración de la tolerancia a la glucosa, anorexia. Poco frecuentes: deshidratación. |
| Alteraciones generales y en el lugar de administración Muy frecuentes: reacciones en el lugar de inyección. Frecuentes: astenia. |
| Parámetros de laboratorio Frecuentes: aumento de los niveles de transaminasas. |
| Alteraciones de la piel y del tejido subcutáneo Frecuentes: prurito, erupción cutánea, alopecia. |
| Alteraciones del sistema respiratorio Frecuentes: disnea. |
| Alteraciones del corazón Frecuentes: bradicardia. Poco frecuentes: taquicardia. |
Estudios poscomerciales
Las reacciones adversas durante el período poscomercial se notificaron de forma espontánea y voluntaria, por lo que no siempre es posible determinar con certeza la frecuencia ni la relación causal con la administración del medicamento.
Reacciones adversas al medicamento notificadas en informes espontáneos:
| Alteraciones del sistema sanguíneo y linfático Trombocitopenia. |
| Del sistema inmunológico |
| Enfermedades de la piel y del tejido subcutáneo |
| Alteraciones del sistema hepatobiliar |
| Alteraciones del corazón Aritmias. |
| Estudios |
Descripción de algunas reacciones adversas
Vesícula biliar y reacciones relacionadas
Los análogos de la somatostatina pueden inhibir la contractilidad de la vesícula biliar y reducir la secreción biliar, lo que podría provocar alteraciones en la vesícula biliar o la formación de sedimentos. Se ha notificado la formación de cálculos biliares en un 15-30% de los receptores tratados con Sandostatin® por vía subcutánea en estudios a largo plazo. La frecuencia de enfermedad en la población general (entre 40 y 60 años de edad) oscila entre el 5 y el 20%. La experiencia a largo plazo con Sandostatin® LAR en pacientes con acromegalia o tumores del sistema gastroenteropancreático indica que el tratamiento con Sandostatin® LAR no incrementa la frecuencia de formación de cálculos biliares en comparación con los preparados administrados por vía subcutánea. La formación de cálculos biliares suele ser asintomática; el tratamiento sintomático de los cálculos incluye la disolución en la vesícula biliar y la intervención quirúrgica.
Alteraciones del tubo digestivo
En casos aislados, las reacciones adversas gastrointestinales pueden simular una obstrucción intestinal aguda, incluyendo distensión abdominal progresiva, dolor epigástrico intenso, dolor abdominal y tensión muscular.
Se sabe que con la continuación del tratamiento disminuye la frecuencia de las reacciones adversas gastrointestinales.
Reacciones de hipersensibilidad y reacciones anafilácticas
Durante el periodo poscomercialización se han notificado reacciones de hipersensibilidad y reacciones alérgicas, que afectan principalmente a la piel y, raramente, a la cavidad bucal y al sistema respiratorio. Se han notificado casos aislados de shock anafiláctico.
Reacciones en el sitio de inyección
Los pacientes que recibieron Sandostatin® LAR generalmente notificaron reacciones en el sitio de inyección como dolor, enrojecimiento, hemorragia, picazón, hinchazón o endurecimiento; en la mayoría de los casos no fue necesaria ninguna atención médica.
Alteraciones del metabolismo y de la nutrición
Aunque la excreción de grasa en las heces puede aumentar, hasta la fecha no hay evidencia de que el tratamiento prolongado con octreótido pueda provocar deficiencias nutricionales debido a malabsorción.
Alteraciones del páncreas
Se han notificado casos muy raros de pancreatitis aguda que se desarrolló en las primeras horas o días tras la administración subcutánea de Sandostatin® y que desapareció tras la suspensión del fármaco. Además, durante la administración prolongada de Sandostatin® por vía subcutánea se han observado casos de pancreatitis asociada a litiasis biliar.
Alteraciones del sistema cardiovascular
La bradicardia es un efecto adverso frecuente en el tratamiento con análogos de la somatostatina. En estudios de ECG en pacientes con acromegalia y en pacientes con síndrome carcinoide durante el tratamiento con el fármaco se han observado: alargamiento del intervalo QT, desviación del eje eléctrico del corazón, repolarización precoz, patrón de bajo voltaje en el ECG, desplazamiento de la zona de transición, onda R precoz y cambios inespecíficos del segmento ST y de la onda T. Dado que muchos de estos pacientes padecen enfermedades cardíacas, no se ha establecido una relación causal entre el desarrollo de estos fenómenos y el acetato de octreótido.
Trombocitopenia
Durante el periodo poscomercialización se han notificado casos de trombocitopenia, especialmente durante el tratamiento con Sandostatin® (vía intravenosa) en pacientes con cirrosis hepática, así como durante el tratamiento con Sandostatin® LAR. Se considera que este fenómeno es reversible tras la interrupción del tratamiento.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite continuar con la vigilancia de la relación beneficio-riesgo del medicamento.
Duración del producto. 3 años.
No utilizar el medicamento después de la fecha de caducidad indicada en el envase.
Condiciones de conservación.
Conservar en el envase original a una temperatura de 2-8 °C.
Conservar en un lugar fuera del alcance de los niños.
Sandostatin® LAR puede mantenerse el día de la inyección a una temperatura inferior a 25 °C.
La suspensión debe prepararse exclusivamente inmediatamente antes de la inyección intramuscular.
Incompatibilidades.
Debido a la falta de estudios de compatibilidad, Sandostatin® LAR no debe mezclarse con otros medicamentos.
Envase.
Polvo en forma de microesferas en un frasco para inyecciones de 6 ml de vidrio incoloro, cerrado con un tapón de goma de color gris con revestimiento de policaucho fluorado y tapón de aluminio de sistema flip-off de color azul oscuro (para dosis de 10 mg), naranja (para dosis de 20 mg) o rojo oscuro (para dosis de 30 mg).
Jeringa precargada con 3 ml de disolvente de vidrio incoloro con dos tapones de goma de color gris, pistón, mango y tapón protector, un adaptador para frasco en un contenedor de plástico, una aguja.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
- Novartis Pharma Stein AG / Novartis Pharma Stein AG (fabricación del lote);
- Novartis Farmaceutica, S.A. / Novartis Farmaceutica, S.A. (fabricación del lote).
Dirección del fabricante y lugar de actividad.
- Schaffhauserstrasse, 4332 Stein, Suiza / Schaffhauserstrasse, 4332 Stein, Switzerland.
- Gran Vía de les Corts Catalanes 764, Barcelona, 08013, España /
Gran Via de les Corts Catalanes 764, Barcelona, 08013, Spain