Sandostatin® lar
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT SANDOSTATIN® LAR (SANDOSTATIN® LAR®)
Composition:
Active substance: octreotide as octreotide acetate;
1 vial containing 10 mg of powder contains 11.2 mg of octreotide acetate, equivalent to 10 mg of octreotide;
1 vial containing 20 mg of powder contains 22.4 mg of octreotide acetate, equivalent to 20 mg of octreotide;
1 vial containing 30 mg of powder contains 33.6 mg of octreotide acetate, equivalent to 30 mg of octreotide;
Excipients: poly(DL-lactide-co-glycolide), mannite (E 421);
solvent for the preparation of injection suspension:
1 pre-filled syringe with a volume of 2.0 ml contains sodium carmellose (sodium carboxymethylcellulose), mannite (E 421), water for injections, poloxamer 188.
Pharmaceutical form. Powder for suspension for injection supplied with solvent.
Main physicochemical properties. Powder ranging from white to white with a yellowish tint. Solvent: from clear to no more intensely opalescent than the European Pharmacopoeia reference suspension, solution from colorless to slightly yellow or brown.
Pharmacotherapeutic group. Hormones for systemic use (excluding sex hormones and insulin). Pituitary, hypothalamic hormones and their analogues. Hypothalamic hormones. Somatostatin and analogues. Octreotide.
ATC code H01C B02.
Pharmacological Properties
Pharmacodynamics
Octreotide is a synthetic octapeptide, a derivative of the natural hormone somatostatin, with similar pharmacological effects but with a significantly longer duration of action. The drug suppresses pathologically increased secretion of growth hormone (GH), as well as peptides and serotonin produced by the gastroenteropancreatic endocrine system.
Studies in animals have shown that octreotide is a more potent inhibitor of GH, glucagon, and insulin secretion than somatostatin; the drug inhibits GH and glucagon secretion with greater selectivity.
In healthy volunteers, octreotide, similar to somatostatin, suppresses:
- GH secretion stimulated by arginine, physical exercise, and insulin-induced hypoglycemia;
- insulin, glucagon, gastrin, and other peptides of the gastroenteropancreatic endocrine system secreted in response to food intake, as well as insulin and glucagon secretion stimulated by arginine;
- thyrotropin (TSH) secretion stimulated by thyrotropin-releasing hormone (TRH).
Unlike somatostatin, octreotide suppresses GH secretion more than insulin secretion, and its administration is not associated with rebound hypersecretion of hormones (e.g., GH in patients with acromegaly).
In patients with acromegaly, Sandostatin® LAR, a depot formulation of octreotide, can be administered at 4-week intervals, providing sustained therapeutic levels of octreotide in blood serum, consistently reducing GH levels and normalizing serum insulin-like growth factor-1 (IGF-1) concentrations in most patients. In the majority of patients, Sandostatin® LAR markedly reduces clinical manifestations of the disease, such as headache, excessive sweating, paresthesia, fatigue, osteoarthralgia, and carpal tunnel syndrome. In a significant proportion (50%) of previously untreated acromegaly patients with GH-hypersecreting pituitary adenomas, treatment with Sandostatin® LAR resulted in tumor volume reduction by >20%. Additionally, short-term studies indicate that octreotide administration prior to surgical treatment in some patients with pituitary adenoma may lead to tumor shrinkage. However, surgical treatment should not be delayed.
In patients with endocrine-active tumors of the gastrointestinal tract and pancreas, treatment with Sandostatin® LAR provides sustained control over various disease-related clinical symptoms. The effects of octreotide on different types of endocrine-active tumors of the gastrointestinal tract and pancreas are described below.
Carcinoid Tumors
Administration of octreotide may lead to symptom relief, particularly flushing and diarrhea. In many cases, this is accompanied by a reduction in plasma serotonin concentration and decreased urinary excretion of 5-hydroxyindoleacetic acid (5-HIAA).
Tumors characterized by vasoactive intestinal peptide (VIP) hyperproduction
The biochemical hallmark of these tumors is excessive VIP synthesis. In most cases, octreotide treatment leads to a reduction in the severity of secretory diarrhea typical of this condition, thereby improving the patient's quality of life. This is associated with a reduction in related electrolyte imbalances, such as hypokalemia, allowing discontinuation of enteral and parenteral fluid and electrolyte replacement. In some patients, computed tomography shows slowed or halted tumor progression and even tumor shrinkage, particularly of liver metastases. Clinical improvement is usually accompanied by a decrease (down to normal levels) in plasma VIP concentration.
Glucagonomas
Octreotide administration leads, in most cases, to a significant reduction in necrolytic migratory erythema, which is characteristic of this condition. Octreotide has no significant effect on the severity of diabetes mellitus, frequently observed in glucagonoma, and usually does not reduce the need for insulin or oral antidiabetic agents. In patients with diarrhea, octreotide reduces its severity, accompanied by an increase in body weight.
With octreotide use, a rapid decrease in plasma glucagon concentration is often observed, although this effect does not persist during long-term treatment. Nevertheless, symptomatic improvement remains stable over a prolonged period.
Gastrinomas / Zollinger–Ellison Syndrome
Therapy with proton pump inhibitors or histamine H2-receptor blockers primarily suppresses gastric hydrochloric acid hypersecretion. However, proton pump inhibitors or H2-receptor blockers do not always adequately alleviate diarrhea, which is also an important symptom of the disease. In some patients, Sandostatin® LAR contributes to further reduction in gastric acid hypersecretion and clinical improvement, including diarrhea, by reducing elevated gastrin levels.
Insulinomas
Octreotide administration reduces immunoreactive insulin levels in the blood. In patients with resectable tumors, octreotide can restore and maintain normoglycemia in the preoperative period. In patients with unresectable benign and malignant tumors, glycemic control may improve even without sustained long-term reduction in blood insulin levels.
Metastatic neuroendocrine intestinal tumors or primary tumors of unknown origin, when other primary sites, except the intestine, have been excluded
A randomized, double-blind, placebo-controlled phase III trial (PROMID) demonstrated that Sandostatin® LAR inhibits tumor growth in patients with metastatic midgut neuroendocrine tumors (small intestine, appendix, cecum, and ascending colon). A total of 85 patients were randomized to receive either Sandostatin® LAR 30 mg every 4 weeks (n = 42) or placebo (n = 43) for up to 18 months or until tumor progression or death.
Key inclusion criteria were: no prior treatment, histologically confirmed diagnosis, locally unresectable or metastatic well-differentiated tumors; functionally active or inactive neuroendocrine tumors/carcinomas; primary tumor located in the midgut (small intestine, appendix, cecum, ascending colon) or of unknown primary origin attributed to the midgut, provided primary sites in the pancreas, thorax, or other locations were excluded.
The primary endpoint was time to tumor progression or tumor-related death (TTP).
An intent-to-treat (ITT) analysis of all randomized patients showed 26 events of progression or tumor-related deaths in the Sandostatin® LAR group versus 41 in the placebo group (HR = 0.32; 95% CI: 0.19–0.55; p = 0.000015).
In a conservative ITT analysis, excluding 3 patients randomized in error, 26 and 40 events of progression or tumor-related death were observed in the Sandostatin® LAR and placebo groups, respectively (HR = 0.34; 95% CI: 0.20–0.59; p-value = 0.000072; figure).
In the conservative ITT (cITT) analysis population, with 3 patients excluded at randomization, 26 and 40 events of tumor progression or tumor-related death were recorded in the Sandostatin® LAR and placebo groups, respectively.
Median time to tumor progression was 14.3 months (95% CI: 11.0–28.8 months) in the Sandostatin® LAR group and 6.0 months (95% CI: 3.7–9.4 months) in the placebo group.
In the per-protocol population analysis, with additional patients excluded at the end of study therapy, 19 and 38 events of tumor progression or tumor-related death were observed in the Sandostatin® LAR and placebo groups, respectively (HR = 0.24; 95% CI: 0.13–0.45; p-value = 0.0000036).
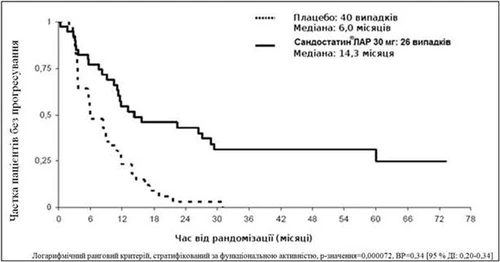
Figure. Kaplan–Meier estimate of time to tumor progression in Sandostatin® LAR and placebo groups (conservative ITT analysis population)
Table 1
Time to tumor progression parameters based on population analyses
| Analysis |
TTP Cases |
Median TTR, months [95% CI] |
HR [95% CI], p* |
||
| Sandostatin® LAR |
Placebo |
Sandostatin® LAR |
Placebo |
||
| ITT |
26 |
41 |
ND |
ND |
0.32 [95% CI: 0.19–0.55] p=0.000015 |
| cITT |
26 |
40 |
14.3 [95% CI: 11.0–28.8] |
6.0 [95% CI: 3.7–9.4] |
0.34 [95% CI: 0.20–0.59] p=0.000072 |
| ND – no data; HR – hazard ratio, TTP – time to tumor progression, ITT – intent-to-treat analysis; cITT – conservative ITT analysis; *log-rank test, stratified by functional status |
|||||
The effect of therapy was similar in patients with functionally active (HR=0.23; 95% CI: 0.09–0.57) and inactive tumors (HR=0.25; 95% CI: 0.10 – 0.59). After 6 months of treatment, stable disease was observed in 66% of patients in the Sandostatin® LAR group and in 37% of patients in the placebo group.
Due to the demonstrated significant clinical efficacy of Sandostatin® LAR established in this pre-planned interim analysis, enrollment of study participants was terminated.
The safety profile of Sandostatin® LAR in this study was consistent with the established safety profile.
Pharmacokinetics.
After a single intramuscular injection of Sandostatin® LAR, a transient initial peak serum concentration of octreotide is achieved within 1 hour after administration, followed by a decline within 24 hours to an undetectable level. After this initial peak on Day 1, in most patients the octreotide concentration remains at a subtherapeutic level over the next 7 days. Thereafter, octreotide concentration rises again, reaching a plateau around Day 14 and remaining at this level for the subsequent 3 – 4 weeks. The peak concentration on Day 1 is lower than the plateau phase concentrations and accounts for no more than 0.5% of the total drug release on Day 1. Approximately 42 days after administration, the octreotide concentration slowly declines, paralleling the terminal degradation phase of the polymer matrix of the dosage form.
In patients with acromegaly, the plateau phase concentration of octreotide after a single injection of Sandostatin® LAR at doses of 10, 20, or 30 mg was 358, 926, and 1710 ng/L, respectively. The steady-state serum concentration of octreotide achieved after 3 injections given at 4-week intervals is 1.6 to 1.8 times higher, reaching up to 1557 ng/L and 2384 ng/L after repeated administration of 20 mg and 30 mg of Sandostatin® LAR, respectively.
In patients with carcinoid tumors, the arithmetic mean (and median) steady-state serum concentrations of octreotide after administration of 10, 20, and 30 mg doses of Sandostatin® LAR were 1231 (894), 2620 (2270), and 3928 (3010) ng/L, respectively.
There was no accumulation of octreotide as would be expected from overlapping release profiles during 28-day injections of Sandostatin® LAR.
The pharmacokinetic profile of octreotide after Sandostatin® LAR injection reflects the release from the polymer matrix and its degradation. Immediately after release into systemic circulation, octreotide distributes according to its pharmacokinetic properties as described for subcutaneous administration. The volume of distribution of octreotide at steady state is 0.27 L/kg, and total clearance is 160 mL/min. Plasma protein binding is approximately 65%, while binding to blood cells is negligible.
Clinical characteristics.
Indications.
Acromegaly (in patients with prior experience of treatment with Sandostatin®):
- treatment of the main manifestations of the disease in cases where surgical treatment or radiotherapy have not provided sufficient efficacy;
- treatment between courses of radiotherapy until its effectiveness is achieved.
Symptoms of functional endocrine tumors of the gastrointestinal tract (in patients with prior experience of treatment with Sandostatin®):
- carcinoid tumors with present carcinoid syndrome;
- VIPomas;
- glucagonomas;
- gastrinomas / Zollinger–Ellison syndrome;
- insulinomas, for control of hypoglycemia in the preoperative period, as well as for maintenance therapy;
- somatoliberinomas (tumors producing growth hormone-releasing factor).
Metastatic neuroendocrine tumors of intestinal origin or tumors of unknown primary site, when other primary locations except intestine have been excluded.
Contraindications.
Hypersensitivity to octreotide or to any of the excipients of the medicinal product (including the solvent).
Interaction with other medicinal products and other forms of interaction.
Dosage adjustment of medicinal products such as beta-blockers, calcium channel blockers, or agents for fluid and electrolyte balance control may be required during concomitant treatment with Sandostatin® LAR.
Dosage adjustment of insulin and antidiabetic medicinal products may be required during concomitant treatment with Sandostatin® LAR.
Octreotide has been shown to reduce intestinal absorption of cyclosporine and to delay absorption of cimetidine.
Concomitant administration of octreotide and bromocriptine increases the bioavailability of bromocriptine.
Limited published data suggest that somatostatin analogs may reduce the metabolic clearance of substances metabolized by cytochrome P450 enzymes, possibly due to inhibition of GH secretion. Since such an effect of octreotide cannot be excluded, caution should be exercised when using other drugs that are primarily metabolized by CYP3A4, as well as those with a narrow therapeutic index (e.g., quinidine, terfenadine).
Special precautions for use.
General
Since pituitary tumors secreting GH may occasionally increase in size, causing serious complications (e.g., visual field defects), careful monitoring of all patients is essential. If signs of tumor enlargement occur, alternative treatment options should be considered.
The therapeutic effect of lowering GH levels and normalizing IGF-1 concentrations in women with acromegaly may potentially restore fertility. Women of childbearing potential should be advised to use appropriate contraceptive methods during treatment with octreotide, if necessary.
Thyroid function should be monitored in patients receiving long-term octreotide therapy.
Liver function should be monitored during octreotide therapy.
Cardiovascular effects
Bradycardia has been frequently reported. Dose adjustments of concomitant medications such as beta-blockers, calcium channel blockers, or drugs affecting fluid or electrolyte balance may be necessary.
Cases of atrioventricular block (including complete atrioventricular block) have been observed in patients receiving high-dose continuous infusion (100 micrograms/hour) and in those receiving intravenous bolus octreotide (50 micrograms bolus followed by 50 micrograms/hour continuous infusion). Therefore, the maximum dose of 50 micrograms/hour should not be exceeded (see section "Dosage and administration"). Patients receiving high-dose intravenous octreotide should be under appropriate cardiac monitoring.
Gallbladder-related effects
Cholelithiasis is the most common complication of Sandostatin® therapy and may be associated with cholecystitis and gallbladder wall thickening (see section "Adverse reactions"). In addition, cases of cholangitis as a complication of cholelithiasis have been reported in patients treated with Sandostatin® LAR during the post-marketing period. Ultrasound examination of the gallbladder is recommended before initiating treatment with Sandostatin® LAR and approximately every 6 months thereafter.
Pancreatic function
Exocrine pancreatic insufficiency (EPI) has been observed in some patients receiving octreotide therapy for gastroenteropancreatic neuroendocrine tumors. Symptoms of EPI may include steatorrhea, diarrhea, abdominal bloating, and weight loss. Screening and appropriate management of EPI according to clinical guidelines should be considered in symptomatic patients.
Glucose metabolism
Due to suppression of GH and inhibition of glucagon and insulin release, Sandostatin® LAR may alter glucose regulation. Postprandial glucose tolerance may be impaired. In some cases, persistent hyperglycemia may develop with long-term administration, as observed in patients receiving subcutaneous Sandostatin®. Hypoglycemia may also occur.
In patients with concomitant type 1 diabetes mellitus, Sandostatin® LAR may affect glucose regulation, reducing insulin requirements. In non-diabetic patients and in patients with type 2 diabetes with partially preserved insulin reserve, subcutaneous Sandostatin® may lead to increased postprandial hyperglycemia. Therefore, monitoring of glucose tolerance and antidiabetic therapy is recommended.
Since octreotide has a relatively greater inhibitory effect on GH and glucagon compared to insulin, and due to its shorter inhibitory effect on insulin secretion, octreotide may increase the severity and duration of hypoglycemia in patients with insulinoma. These patients should be closely monitored.
Nutrition
Octreotide may impair absorption of dietary fats in some patients.
Decreased vitamin B12 levels and abnormal Schilling test results have been observed in some patients receiving octreotide therapy. Vitamin B12 levels should be monitored in patients with a history of vitamin B12 deficiency during treatment with Sandostatin® LAR.
Sodium content
Sandostatin® LAR contains less than 1 mmol (23 mg) of sodium per dose and may therefore be considered sodium-free.
Use during pregnancy or breastfeeding
Data on the use of octreotide in pregnant women are limited (fewer than 300 pregnancy outcomes reported), and pregnancy outcomes are unknown in approximately one-third of cases. Most reports were obtained from post-marketing use of octreotide, and the proportion of pregnant women among female patients with acromegaly exceeded 50%. Most women received octreotide during the first trimester at doses of 100–1200 micrograms/day as subcutaneous Sandostatin® or 10–40 mg/month of Sandostatin® LAR. Congenital anomalies were reported in approximately 4% of pregnancies with known outcomes. No causal relationship with octreotide was suspected in these cases.
Animal studies have not shown direct or indirect harmful effects with regard to reproductive toxicity.
As a precautionary measure, use of Sandostatin® during pregnancy should be avoided.
Breastfeeding
It is unknown whether octreotide passes into human breast milk. Animal studies have shown excretion of octreotide into breast milk. Women receiving treatment with Sandostatin® LAR should discontinue breastfeeding.
Fertility
It is unknown whether octreotide affects human fertility. In newborn male infants exposed to the drug during pregnancy and breastfeeding, delayed testicular descent has been observed. However, in experimental studies in male and female rats, octreotide did not affect fertility at doses up to 1 mg/kg body weight per day.
Ability to influence reaction speed when driving or operating machinery
Sandostatin® LAR has no or negligible influence on the ability to drive or operate machinery. Patients should be advised to exercise caution when driving a car or operating machinery if they experience dizziness, asthenia/fatigue, or headache during treatment with Sandostatin® LAR.
Administration and Dosage
Acromegaly
Treatment with Sandostatin® LAR is recommended to begin with 20 mg administered every 4 weeks for a 3-month period. Patients currently receiving Sandostatin® may initiate treatment with Sandostatin® LAR the day after their last subcutaneous Sandostatin® injection. Subsequent dose adjustments should be based on plasma growth hormone (GH) levels, insulin-like growth factor-1 (IGF-1) concentrations, and clinical signs of the disease.
For patients in whom clinical manifestations and biochemical parameters (GH, IGF-1) are not adequately controlled within this 3-month period (GH concentrations remain above 2.5 µg/L), the dose may be increased to 30 mg every 4 weeks. If, after a 3-month treatment period with 30 mg, GH and IGF-1 concentrations and/or symptoms remain inadequately controlled, the dose may be increased to 40 mg every 4 weeks.
For patients in whom GH concentrations remain below 1 µg/L after 3 months of 20 mg treatment, IGF-1 levels in serum have normalized, and previous symptoms of acromegaly have resolved, the dose may be reduced to 10 mg of Sandostatin® LAR every 4 weeks. However, in such patients receiving a lower dose of Sandostatin® LAR, close monitoring of GH and IGF-1 concentrations and clinical symptoms is recommended.
Assessment of GH and IGF-1 concentrations in patients receiving a stable dose of Sandostatin® LAR should be performed every 6 months.
Gastrointestinal and Pancreatic Endocrine Tumors
Treatment of patients with symptoms related to hormonally active gastrointestinal and pancreatic neuroendocrine tumors
Treatment is recommended to begin with 20 mg of Sandostatin® LAR administered every 4 weeks for 3 months. During the first 2 weeks after the initial Sandostatin® LAR injection, subcutaneous administration of Sandostatin® should continue at the previously effective dose.
For patients in whom symptoms and biological markers are adequately controlled after 3 months of treatment, the dose of Sandostatin® LAR may be reduced to 10 mg every 4 weeks.
For patients in whom symptoms and biological markers are only partially controlled after 3 months of treatment, the dose may be increased to 30 mg every 4 weeks.
On days when symptoms related to gastrointestinal or pancreatic tumors worsen during Sandostatin® LAR treatment, additional subcutaneous administration of Sandostatin® at the dose previously used before initiating Sandostatin® LAR therapy is recommended. This may occur primarily during the first 2 months of treatment, until therapeutic concentrations of octreotide are achieved.
Treatment of patients with metastatic midgut neuroendocrine tumors or with unknown primary tumor site when other primary locations, except midgut, have been excluded
The recommended dose of Sandostatin® LAR is 30 mg once every 4 weeks. Sandostatin® LAR therapy for tumor control should be continued even in the absence of disease progression.
Use in patients with renal impairment
Renal impairment does not affect the overall exposure to octreotide when administered as subcutaneous Sandostatin®. Therefore, no dose adjustment of Sandostatin® LAR is necessary in these patients.
Use in patients with hepatic impairment
Studies with subcutaneous and intravenous administration of Sandostatin® have shown that drug elimination may be reduced in patients with liver cirrhosis, but not in patients with hepatic steatosis. In some cases, dose adjustment of Sandostatin® LAR may be required in patients with hepatic impairment.
Use in elderly patients
Studies with subcutaneous administration of Sandostatin® have shown that dose adjustment is not required in patients aged 65 years and older.
Administration method
Sandostatin® LAR is intended for long-term treatment as directed by a physician.
To minimize injection site pain, it is recommended to allow the Sandostatin® LAR medication to reach room temperature before administration.
To prepare the Sandostatin® LAR suspension and administer deep intramuscular injection into the gluteal muscle, the components of the injection kit should be used.
Sandostatin® LAR is administered as a deep intramuscular injection. With repeated administration, the injection site should be alternated between the right and left gluteal muscles.
Instructions for administering intramuscular injection of Sandostatin® LAR
for deep intramuscular injection into the gluteal muscle only:
Package contents:
**
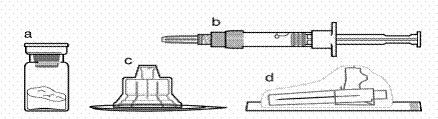
**
a one vial containing Sandostatin® LAR medication
b one pre-filled syringe with solvent
c one vial adapter in a plastic container
d one safety injection needle (0.91 mm × 38.1 mm; 19G × 1.5") in a protective cap
| Follow carefully the instructions below to properly prepare Sandostatin® LAR for deep intramuscular administration. Sandostatin® LAR suspension must be prepared immediately before use. The injection must be administered only by an experienced healthcare professional. |
|
|
|
Step 1 Remove Sandostatin® LAR from the refrigerator and ensure it has reached room temperature. This usually takes between 30 and 60 minutes, but no longer than 24 hours. Wash your hands with soap and warm water. Place the kit on a clean, flat surface. Remove the lid film from the tray containing the injection set. Remove the cap from the vial containing Sandostatin® LAR. |
|
|
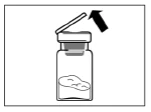
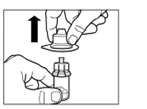
Step 2 Wipe the rubber stopper of the vial with an alcohol swab. Caution: do not touch the rubber stopper after it has been wiped with alcohol. Remove the protective film from the plastic container holding the vial adapter. DO NOT remove the adapter from the plastic container. Place the plastic container with the adapter on top of the vial and press downward until you hear a distinct clicking sound. Remove the plastic container vertically upward from the vial adapter. |
|
|
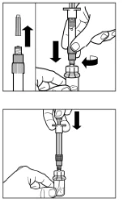
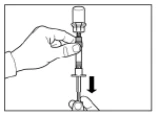
Step 3 Remove the cap from the syringe containing the solvent and screw the syringe into the vial adapter. Slowly push the plunger fully down to transfer all the solvent into the vial. |
|
|
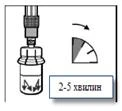
Step 4 The vial should remain standing in an upright position until the solvent has completely wet the Sandostatin® LAR powder (at least 2–5 minutes). During this time, prepare the patient for injection. Note: It is normal for the plunger of the syringe to rise slightly due to a slight increase in pressure within the vial. |
|
|
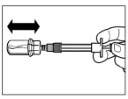
Step 5 After dissolution, press the syringe plunger down again fully and hold it in place. Gently shake the vial horizontally for approximately 30 seconds to mix the contents. Visually confirm that the powder has completely dissolved (a uniform milky suspension should form). If the powder has not fully dissolved, continue shaking the vial for another 30 seconds. |
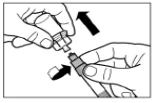
|
|
Step 6 Position the syringe and vial upside down and slowly pull back on the plunger to transfer the contents from the vial into the syringe. Unscrew the syringe from the vial adapter. The reconstituted preparation must be administered immediately after preparation. |
|
|
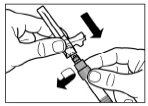
Step 7 Screw the needle onto the syringe. Remove the protective needle cap by pulling it off. Carefully invert the syringe, maintaining homogeneity of its contents. Gently tap the syringe with your finger to remove any visible air bubbles. The reconstituted Sandostatin® LAR solution is ready for immediate administration. |
|
|
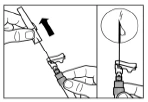
Step 8 Sandostatin® LAR must be administered only by deep intramuscular injection. Intravenous administration is contraindicated. Clean the injection site with an alcohol swab. Fully insert the needle into the right or left gluteal muscle. Gently pull back on the syringe plunger to ensure the needle has not entered a blood vessel; if it has, reposition the needle. Slowly depress the syringe plunger to administer the required dose. After completing the injection, withdraw the needle from the injection site and activate the safety mechanism as shown below. |
|
|
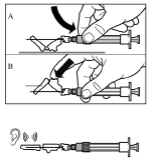
Step 9 Activate the needle safety mechanism with one hand using one of the methods described below: A. Press the hinged part of the safety device against a hard surface, such as a table. B. Press the hinged part of the safety device with your index finger. Remember to keep your fingers behind the needle cannula at all times. A clicking sound confirms proper activation of the safety mechanism. Immediately dispose of the vial, syringe, and needle into a sharps container or another rigid, closed container for safe disposal. |
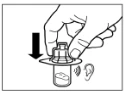
Children.
Somatostatin® LAR should not be used in children due to lack of clinical experience.
Overdose.
A limited number of accidental overdoses of Somatostatin® LAR (dose range 100–163 mg/month) have been reported. The only adverse event reported was flushing. There have been reports of cancer patients receiving Somatostatin® LAR doses up to 60 mg/month and up to 90 mg/2 weeks. These doses were generally well tolerated. However, adverse events such as frequent urination, fatigue, depression, anxiety, and loss of concentration have been observed.
Treatment is symptomatic.
Adverse Reactions
Summary of the drug's safety profile
The most common adverse reactions associated with octreotide treatment involve the gastrointestinal tract, nervous system, liver and gallbladder, and metabolism and nutrition.
The most frequently reported adverse reactions during clinical trials with octreotide were diarrhea, abdominal pain, nausea, flatulence, headache, cholelithiasis, hyperglycemia, and constipation. Other common adverse reactions included dizziness, injection site pain, gallstones, thyroid dysfunction (e.g., decreased levels of TSH, decreased total T4 and free T4), loose stools, impaired glucose tolerance, vomiting, asthenia, and hypoglycemia.
Occasionally, gastrointestinal adverse effects may mimic acute intestinal obstruction with progressive abdominal distension, severe epigastric pain, tenderness, and abdominal muscle rigidity.
Although fecal fat excretion may increase, there is no evidence that long-term treatment with Sandostatin® LAR leads to nutritional deficiencies due to malabsorption.
Acute pancreatitis has been reported very rarely, occurring within the first hours or days of Sandostatin® LAR treatment and resolving upon discontinuation of the drug. In addition, cases of pancreatitis secondary to cholelithiasis have been reported in patients receiving long-term Sandostatin® LAR therapy.
ECG changes such as QT interval prolongation, axis deviation, early repolarization, low voltage, R/S transition, early R wave progression, and non-specific ST-T wave changes have been observed in patients with acromegaly as well as in those with carcinoid syndrome during octreotide therapy. A causal relationship between these findings and octreotide has not been established, as many of these patients have underlying cardiovascular disease.
The adverse reactions listed below were observed during clinical trials of octreotide and are categorized by frequency: very common (≥ 1/10); common (≥ 1/100, < 1/10); uncommon (≥ 1/1,000, < 1/100); rare (≥ 1/10,000, < 1/1,000); very rare (< 1/10,000), including isolated case reports. Within each frequency category, adverse reactions are listed in order of decreasing severity.
Adverse reactions reported during clinical studies
| Disorders of the gastrointestinal system Very common: diarrhea, abdominal pain, nausea, constipation, flatulence. Common: dyspepsia, vomiting, bloating, steatorrhea, frequent loose stools, discoloration of feces. |
| Disorders of the nervous system Very common: headache. Common: dizziness. |
| Disorders of the endocrine system Common: hypothyroidism, thyroid function abnormalities (e.g. decreased TSH levels, decreased total T4 levels, decreased free T4 levels). |
| Disorders of the hepatobiliary system Very common: cholelithiasis. Common: cholecystitis, gallstones, hyperbilirubinemia. |
| Metabolism and nutrition disorders Very common: hyperglycemia. Common: hypoglycemia, impaired glucose tolerance, anorexia. Uncommon: dehydration. |
| General disorders and administration site conditions Very common: injection site reactions. Common: asthenia. |
| Laboratory findings Common: increased transaminase levels. |
| Disorders of the skin and subcutaneous tissue Common: pruritus, rash, alopecia. |
| Disorders of the respiratory system Common: dyspnea. |
| Cardiac disorders Common: bradycardia. Uncommon: tachycardia. |
Post-marketing studies
Adverse reactions reported during the post-marketing period were reported spontaneously and voluntarily, and therefore it is not always possible to reliably determine their frequency or establish a causal relationship with the use of the medicinal product.
Adverse reactions reported in spontaneous reports:
| Blood and lymphatic system disorders Thrombocytopenia. |
| Immune system disorders |
| Skin and subcutaneous tissue disorders |
| Hepatobiliary disorders |
| Cardiac disorders Arrhythmias. |
| Investigations |
Description of some adverse reactions
Gallbladder and related reactions
Somatostatin analogues may inhibit gallbladder contractility and reduce bile secretion, which may lead to gallbladder abnormalities or sludge formation. Gallstone formation has been reported in 15–30% of recipients receiving subcutaneous Sandostatin® in long-term studies. The prevalence in the general population (aged 40 to 60 years) ranges from 5 to 20%. Long-term exposure to Sandostatin® LAR in patients with acromegaly or gastroenteropancreatic tumors indicates that treatment with Sandostatin® LAR does not increase the incidence of gallstone formation compared to treatment with subcutaneously administered formulations. Gallstone formation is usually asymptomatic; symptomatic management includes dissolution in the gallbladder and surgical intervention.
Gastrointestinal disorders
In isolated cases, gastrointestinal adverse reactions may mimic acute intestinal obstruction, including progressive abdominal distension, severe epigastric pain, abdominal tenderness, and muscle rigidity.
It is known that with continued treatment, the frequency of gastrointestinal adverse reactions decreases.
Hypersensitivity and anaphylactic reactions
During the post-marketing period, hypersensitivity and allergic reactions have been reported, primarily affecting the skin, and rarely the oral cavity and respiratory tract. Isolated cases of anaphylactic shock have been reported.
Injection site reactions
Patients receiving Sandostatin® LAR commonly reported the following injection site reactions: pain, redness, bleeding, itching, swelling, or induration; in most cases, no medical intervention was required.
Metabolism and nutrition disorders
Although fecal fat excretion may increase, there is currently no evidence that long-term treatment with octreotide leads to nutrient deficiency due to malabsorption.
Pancreatic disorders
Very rare cases of acute pancreatitis have been reported, occurring within the first hours or days after subcutaneous administration of Sandostatin® and resolving upon discontinuation of the drug. Additionally, cases of pancreatitis associated with cholelithiasis have been observed during prolonged subcutaneous administration of Sandostatin®.
Cardiovascular disorders
Bradycardia is a common adverse effect during treatment with somatostatin analogues. ECG studies in patients with acromegaly and patients with carcinoid syndrome treated with the drug have shown QT interval prolongation, deviation of the cardiac axis, early repolarization, low-voltage ECG pattern, shift of the transitional zone, early R wave, and non-specific ST segment and T wave changes. Since many of these patients have underlying heart disease, a causal relationship between these findings and octreotide acetate has not been established.
Thrombocytopenia
During the post-marketing period, thrombocytopenia has been reported, particularly during treatment with Sandostatin® (intravenous) in patients with liver cirrhosis, as well as during treatment with Sandostatin® LAR. This phenomenon is considered reversible upon discontinuation of treatment.
Reporting suspected adverse reactions
Reporting suspected adverse reactions after drug authorization is important. It enables continuous monitoring of the benefit-risk balance of the medicinal product.
Shelf life. 3 years.
Do not use the medicinal product after the expiry date stated on the packaging.
Storage conditions.
Store in the original packaging at 2–8°C.
Keep out of the reach of children.
On the day of injection, Sandostatin® LAR may be kept at a temperature below 25°C.
The suspension should be prepared exclusively immediately before intramuscular injection.
Incompatibilities.
Due to the lack of compatibility studies, Sandostatin® LAR must not be mixed with other medicinal products.
Packaging.
Powder as microspheres in a 6 ml injection vial made of colorless glass, closed with a grey rubber stopper with fluorocarbon coating and a dark blue (for 10 mg dosage), orange (for 20 mg dosage), or dark red (for 30 mg dosage) flip-off aluminum cap.
Prefilled syringe with 3 ml solvent made of colorless glass, with two grey rubber stoppers, plunger, barrel, cap, one vial adapter in a plastic container, one needle.
Prescription category.
Prescription only.
Manufacturer.
- Novartis Pharma Stein AG / Novartis Pharma Stein AG (batch release)
- Novartis Farmaceutica, S.A. / Novartis Farmaceutica, S.A. (batch release)
Manufacturer's address and location of operations.
- Schaffhauserstrasse, 4332 Stein, Switzerland
- Gran Via de les Corts Catalanes 764, Barcelona, 08013, Spain