Lyrica
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO LYRICA® (LYRICA)
Composición:
Principio activo: pregabalina;
1 cápsula contiene 50 mg, 75 mg, 150 mg o 300 mg de pregabalina;
Sustancias auxiliares: lactosa monohidrato; almidón de maíz; talco; envoltura de la cápsula: gelatina, agua, dióxido de titanio (E 171), laurilsulfato de sodio, dióxido de silicio coloidal anhidro, óxido de hierro rojo (E 172) (para cápsulas de 75 mg y 300 mg); tinta: laca, óxido de hierro negro (E 172), propilenglicol, hidróxido de potasio.
Forma farmacéutica. Cápsulas.
Propiedades físico-químicas principales.
- Cápsulas de 50 mg: cápsulas duras opacas (blanco/blanco) de gelatina, tamaño 3, que contienen polvo de color blanco o casi blanco. Impresión en el cuerpo "PGN 50", en la tapa "Pfizer", con tinta negra.
- Cápsulas de 75 mg: cápsulas duras opacas (blanco/naranja) de gelatina, tamaño 4, que contienen polvo de color blanco o casi blanco. Impresión en el cuerpo "PGN 75", en la tapa "Pfizer", con tinta negra.
- Cápsulas de 150 mg: cápsulas duras opacas (blanco/blanco) de gelatina, tamaño 2, que contienen polvo de color blanco o casi blanco. Impresión en el cuerpo "PGN 150", en la tapa "Pfizer", con tinta negra.
- Cápsulas de 300 mg: cápsulas duras opacas (blanco/naranja) de gelatina, tamaño 0, que contienen polvo de color blanco o casi blanco. Impresión en el cuerpo "PGN 300", en la tapa "Pfizer", con tinta negra.
Grupo farmacoterapéutico. Agentes antiepilépticos. Otros agentes antiepilépticos.
Código ATC N03A X16.
Propiedades farmacodinámicas.
Farmacodinámica.
Sustancia activa: pregabalina, análogo del ácido gamma-aminobutírico [(S)-3-(aminometil)-5-metilhexanoico].
Mecanismo de acción.
La pregabalina se une a la subunidad auxiliar (proteína α2-δ) de los canales de calcio dependientes del potencial en el sistema nervioso central (SNC).
Eficacia clínica y seguridad.
- Dolor neuropático.
Durante los estudios se demostró la eficacia del medicamento para el tratamiento de la neuropatía diabética, neuralgia posherpética y lesión de la médula espinal. No se ha estudiado la eficacia del medicamento en otros tipos de dolor neuropático.
La pregabalina fue evaluada en 10 ensayos clínicos controlados de hasta 13 semanas de duración con un régimen de dosificación dos veces al día, y en estudios de hasta 8 semanas con un régimen de tres veces al día. En general, los perfiles de seguridad y eficacia para los regímenes de dosificación dos y tres veces al día fueron similares.
En ensayos clínicos controlados de hasta 12 semanas de duración, en los que el medicamento se utilizó para tratar el dolor neuropático, la reducción del dolor de origen periférico y central se observó ya en la primera semana y se mantuvo durante todo el período de tratamiento.
En ensayos clínicos controlados de dolor neuropático periférico, el 35 % de los pacientes que recibieron pregabalina y el 18 % de los pacientes que recibieron placebo mostraron una mejoría del 50 % en la escala de evaluación del dolor. Entre los pacientes que no presentaron somnolencia, dicha mejoría se observó en el 33 % de los pacientes que recibieron pregabalina y en el 18 % de los pacientes del grupo placebo. Entre los pacientes que presentaron somnolencia, la proporción de pacientes respondedores fue del 48 % en el grupo de pregabalina y del 16 % en el grupo placebo.
En un ensayo clínico controlado de dolor neuropático de origen central, el 22 % de los pacientes que recibieron pregabalina y el 7 % de los pacientes que recibieron placebo mostraron una mejoría del 50 % en la escala de evaluación del dolor.
- Epilepsia.
Tratamiento adyuvante. La pregabalina fue evaluada en 3 ensayos clínicos controlados de 12 semanas de duración con un régimen de dosificación dos o tres veces al día. En general, los perfiles de seguridad y eficacia para los regímenes de dosificación dos y tres veces al día fueron similares.
La reducción de la frecuencia de crisis convulsivas se observó ya en la primera semana.
Pacientes pediátricos. La eficacia y seguridad de la pregabalina como tratamiento adyuvante en epilepsia en niños menores de 12 años y adolescentes no han sido establecidas. Las reacciones adversas observadas en el estudio de farmacocinética y tolerabilidad, que incluyó pacientes de 3 meses a 16 años de edad (n=65) con crisis parciales, fueron similares a las observadas en adultos. Los resultados de un ensayo controlado con placebo de 12 semanas con 295 niños de 4 a 16 años de edad y un ensayo controlado con placebo de 14 días con 175 niños de 1 mes a menos de 4 años, cuyo objetivo fue evaluar la eficacia y seguridad de la pregabalina como tratamiento adicional de crisis parciales, y dos estudios abiertos de seguridad de 1 año de duración con 54 y 431 niños respectivamente, de 3 meses a 16 años con epilepsia, indican que reacciones adversas como fiebre e infecciones de las vías respiratorias superiores ocurren con mayor frecuencia en niños que en adultos con epilepsia (véanse las secciones «Instrucciones de uso y dosis», «Reacciones adversas» y «Farmacocinética»).
En el ensayo controlado con placebo de 12 semanas, a los niños (de 4 a 16 años) se les administró pregabalina a dosis de 2,5 mg/kg/día (máximo 150 mg/día), pregabalina a dosis de 10 mg/kg/día (máximo 600 mg/día) o placebo. Una reducción de al menos el 50 % de las crisis parciales respecto al valor basal se observó en el 40,6 % de los pacientes que recibieron pregabalina a dosis de 10 mg/kg/día (p=0,0068 frente a placebo), en el 29,1 % de los pacientes que recibieron pregabalina a dosis de 2,5 mg/kg/día (p=0,2600 frente a placebo) y en el 22,6 % de los que recibieron placebo.
En el ensayo controlado con placebo de 14 días, a los niños (de 1 mes a menos de 4 años) se les administró pregabalina a dosis de 7 mg/kg/día, pregabalina a dosis de 14 mg/kg/día o placebo. La frecuencia mediana diaria de crisis fue de 4,7 y 3,8 para pregabalina a dosis de 7 mg/kg/día, 5,4 y 1,4 para pregabalina a dosis de 14 mg/kg/día, y 2,9 y 2,3 para placebo, en los valores basal y final, respectivamente. La pregabalina a dosis de 14 mg/kg/día redujo significativamente la frecuencia de crisis parciales transformada logarítmicamente en comparación con placebo (p = 0,0223); la pregabalina a dosis de 7 mg/kg/día no mostró mejoría frente a placebo.
En un ensayo controlado con placebo de 12 semanas con pacientes con crisis tónico-clónicas generalizadas primarias (CTCG), se administró pregabalina a dosis de 5 mg/kg/día (máximo 300 mg/día) o 10 mg/kg/día (máximo 600 mg/día) o placebo como tratamiento adyuvante a 219 pacientes de 5 a 65 años (66 de ellos de 5 a 16 años). Una reducción de al menos el 50 % de la frecuencia de crisis CTCG se observó en el 41,3 %, 38,9 % y 41,7 % de los pacientes que recibieron pregabalina a dosis de 5 mg/kg/día, pregabalina a dosis de 10 mg/kg/día y placebo, respectivamente.
Monoterapia (en pacientes con enfermedad recién diagnosticada). La pregabalina fue evaluada en un ensayo clínico controlado de 56 semanas con un régimen de dosificación dos veces al día. Al utilizar pregabalina no se alcanzó un nivel de eficacia comparable al de lamotrigina, según la evaluación a los 6 meses con el criterio principal de ausencia de crisis convulsivas. La pregabalina y la lamotrigina fueron igualmente seguras y bien toleradas.
- Trastorno de ansiedad generalizada.
La pregabalina fue evaluada en 6 ensayos controlados de 4–6 semanas de duración, un estudio de 8 semanas con pacientes de edad avanzada y un estudio prolongado de prevención de recaídas con una fase doble ciego de prevención de recaídas de 6 meses.
La reducción de los síntomas del trastorno de ansiedad generalizada según la escala de ansiedad de Hamilton (HAM-A) se observó ya en la semana 1.
En ensayos clínicos controlados (de 4–8 semanas de duración), el 52 % de los pacientes que recibieron pregabalina y el 38 % de los pacientes del grupo placebo mostraron una mejoría de al menos el 50 % en la puntuación total de la escala HAM-A desde el valor basal hasta el punto final.
Durante los ensayos controlados, la visión borrosa se observó con mayor frecuencia en pacientes que recibieron pregabalina que en aquellos que recibieron placebo. En la mayoría de los casos, este fenómeno desapareció con la continuación del tratamiento. Se realizó un examen oftalmológico (incluyendo evaluación de la agudeza visual, evaluación formal del campo visual y examen del fondo de ojo con pupilas dilatadas) en más de 3600 pacientes en el marco de ensayos clínicos controlados. Entre estos pacientes, la agudeza visual empeoró en el 6,5 % en el grupo de pregabalina y en el 4,8 % en el grupo placebo. Cambios en el campo visual se detectaron en el 12,4 % de los pacientes que recibieron pregabalina y en el 11,7 % de los pacientes del grupo placebo. Cambios en el fondo de ojo se detectaron en el 1,7 % de los pacientes que recibieron pregabalina y en el 2,1 % de los pacientes del grupo placebo.
- Fibromialgia.
La eficacia del medicamento Lyrica fue establecida en un estudio multicéntrico doble ciego controlado con placebo de 14 semanas (F1) y en un estudio aleatorizado de retirada de 6 semanas (F2). En estos estudios se incluyeron pacientes con diagnóstico de fibromialgia según los criterios del Colegio Estadounidense de Reumatología (dolor generalizado durante al menos 3 meses y dolor presente en 11 o más de 18 puntos sensibles específicos). Los estudios demostraron una reducción del dolor según la escala analógica visual. Además, se observó mejoría según la evaluación general del paciente y según el cuestionario sobre el impacto de la fibromialgia.
Pacientes pediátricos. Se realizó un estudio controlado con placebo de 15 semanas con 107 niños de 12–17 años con fibromialgia que recibieron Lyrica en dosis de 75–450 mg/día. Según la evaluación del criterio primario de eficacia (cambio en la intensidad general del dolor desde el valor basal hasta la semana 15, calculado mediante una escala numérica de 11 puntos), se observó una mejoría numéricamente mayor en los pacientes que recibieron pregabalina en comparación con los que recibieron placebo, pero esta mejoría no alcanzó significación estadística. Las reacciones adversas más frecuentes observadas en los estudios clínicos fueron mareo, náuseas, cefalea, aumento de peso y fatiga. El perfil general de seguridad en adolescentes fue similar al de adultos con fibromialgia.
Farmacocinética.
Los parámetros farmacocinéticos de la pregabalina en estado de equilibrio fueron similares en voluntarios sanos, pacientes con epilepsia que tomaban medicamentos antiepilépticos y pacientes con dolor crónico.
Absorción.
La pregabalina se absorbe rápidamente en ayunas y alcanza concentraciones máximas en plasma dentro de la primera hora tras una dosis única o múltiple. La biodisponibilidad calculada de la pregabalina tras administración oral es ≥ 90 % e independiente de la dosis. Tras administración múltiple, el estado de equilibrio se alcanza en 24–48 horas. La velocidad de absorción de la pregabalina disminuye cuando se administra con alimentos, lo que provoca una reducción de aproximadamente el 25–30 % en la concentración máxima (Cmax) y un alargamiento del tmax a aproximadamente 2,5 horas. Sin embargo, la administración de pregabalina con alimentos no tuvo un impacto clínicamente significativo sobre el grado de absorción.
Distribución.
En estudios preclínicos se demostró que la pregabalina atraviesa la barrera hematoencefálica en ratones, ratas y monos. Se ha establecido que la pregabalina atraviesa la placenta en ratas y se excreta en la leche de ratas durante la lactancia. En humanos, el volumen de distribución de la pregabalina tras administración oral es de aproximadamente 0,56 l/kg. La pregabalina no se une a las proteínas plasmáticas.
Metabolismo.
En humanos, la pregabalina sufre un metabolismo mínimo. Tras la administración de una dosis marcada radiactivamente, aproximadamente el 98 % de la sustancia radiactiva se excretó en orina como pregabalina inalterada. La fracción del metabolito principal, la derivada N-metilada de la pregabalina, detectada en orina, fue del 0,9 % de la dosis administrada. Durante estudios preclínicos no se produjo racemización del enantiómero S de la pregabalina al enantiómero R.
Eliminación.
La pregabalina se elimina del torrente sanguíneo sistémico sin cambios, principalmente por vía renal. El periodo medio de eliminación de la pregabalina es de 6,3 horas. El aclaramiento plasmático y renal de la pregabalina es directamente proporcional al aclaramiento de creatinina (véase la sección «Farmacocinética. Insuficiencia renal»).
Los pacientes con alteración de la función renal o pacientes en hemodiálisis requieren ajuste de la dosis del medicamento (véase la sección «Instrucciones de uso y dosis», tabla).
Linealidad/no linealidad.
La farmacocinética de la pregabalina es lineal en todo el rango de dosis recomendado. La variabilidad de la farmacocinética de la pregabalina entre pacientes es baja (< 20 %). La farmacocinética tras administración múltiple es predecible a partir de los datos obtenidos tras dosis única. Por lo tanto, no es necesario el control planificado de las concentraciones de pregabalina en plasma.
Sexo.
Los resultados de los estudios clínicos indican ausencia de impacto clínicamente significativo del sexo sobre las concentraciones de pregabalina en plasma.
Insuficiencia renal.
El aclaramiento de la pregabalina es directamente proporcional al aclaramiento de creatinina. Además, la pregabalina se elimina eficazmente del plasma mediante hemodiálisis (tras 4 horas de hemodiálisis, la concentración de pregabalina en plasma se reduce aproximadamente en un 50 %). Dado que el medicamento se elimina principalmente por vía renal, es necesario reducir la dosis en pacientes con insuficiencia renal y administrar una dosis adicional tras hemodiálisis (véase la sección «Instrucciones de uso y dosis», tabla).
Insuficiencia hepática.
No se han realizado estudios farmacocinéticos específicos en pacientes con insuficiencia hepática. Dado que la pregabalina no sufre un metabolismo significativo y se excreta en orina principalmente sin cambios, es poco probable que la alteración de la función hepática tenga un impacto significativo sobre la concentración de pregabalina en plasma.
Pacientes pediátricos.
La farmacocinética de la pregabalina fue evaluada en niños con epilepsia (grupos de edad: de 1 a 23 meses, de 2 a 6 años, de 7 a 11 años y de 12 a 16 años) con dosis de 2,5 mg/kg/día, 5 mg/kg/día, 10 mg/kg/día y 15 mg/kg/día en un estudio de farmacocinética y tolerabilidad.
Tras la administración oral de pregabalina en ayunas a niños, el tiempo para alcanzar la concentración máxima en plasma fue en general similar en todos los grupos de edad y osciló entre 0,5 y 2 horas tras la dosis.
Los valores de Cmax y del área bajo la curva de concentración-tiempo (AUC) de la pregabalina aumentaron linealmente con el incremento de la dosis en cada grupo de edad. En niños con peso corporal inferior a 30 kg, los valores de AUC fueron un 30 % más bajos, debido a un incremento del 43 % en el aclaramiento corregido por peso corporal en estos pacientes en comparación con pacientes con peso corporal ≥ 30 kg.
El periodo final de semieliminación de la pregabalina fue en promedio de aproximadamente 3–4 horas en niños menores de 6 años y de 4–6 horas en niños de 7 años o más.
En un análisis farmacocinético poblacional se demostró que el aclaramiento de creatinina fue una covariable significativa para el aclaramiento oral de pregabalina, y el peso corporal fue una covariable significativa para el volumen aparente de distribución oral de pregabalina, y esta relación fue similar en niños y adultos.
No se ha estudiado la farmacocinética de la pregabalina en pacientes menores de 3 meses (véanse las secciones «Instrucciones de uso y dosis», «Reacciones adversas» y «Farmacodinámica»).
Pacientes de edad avanzada.
El aclaramiento de la pregabalina tiende a disminuir con la edad. Esta reducción del aclaramiento de pregabalina tras administración oral concuerda con la disminución del aclaramiento de creatinina asociada al envejecimiento. Los pacientes con alteración de la función renal relacionada con la edad podrían requerir reducción de la dosis de pregabalina (véase la sección «Instrucciones de uso y dosis», tabla 1).
Lactancia.
La farmacocinética de la pregabalina tras su administración a dosis de 150 mg cada 12 horas (dosis diaria de 300 mg) fue evaluada en 10 mujeres lactantes al menos 12 semanas después del parto. La lactancia no tuvo efecto o tuvo un efecto mínimo sobre la farmacocinética de la pregabalina. La pregabalina pasó a la leche materna, con concentraciones medias en estado de equilibrio de aproximadamente el 76 % de las concentraciones en plasma materno. La dosis calculada que recibe el lactante a través de la leche materna (con un consumo medio de leche de 150 ml/kg/día) de una mujer que toma pregabalina a dosis de 300 mg/día o a la dosis máxima de 600 mg/día es de 0,31 o 0,62 mg/kg/día, respectivamente. Estas dosis calculadas representan aproximadamente el 7 % de la dosis diaria total materna ajustada por kg.
Características clínicas.
Indicaciones.
Neuropatía dolorosa.
El medicamento Lyrica está indicado para el tratamiento del dolor neuropático de origen periférico o central en adultos.
Epilepsia.
El medicamento Lyrica está indicado en adultos como tratamiento adicional de crisis parciales con o sin generalización secundaria.
Trastorno de ansiedad generalizada.
El medicamento Lyrica está indicado para el tratamiento del trastorno de ansiedad generalizada en adultos.
Fibromialgia.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes enumerados en la sección «Composición».
Interacción con otros medicamentos y otras formas de interacción.
Dado que la pregabalina se excreta principalmente inalterada por la orina, sufre un metabolismo mínimo en el organismo humano (≤ 2 % de la dosis se elimina por orina en forma de metabolitos), no inhibe el metabolismo de otros fármacos in vitro y no se une a las proteínas plasmáticas, es poco probable que la pregabalina pueda provocar una interacción farmacocinética o ser objeto de dicha interacción.
Estudios in vivo y análisis farmacocinético poblacional.
Por consiguiente, en estudios in vivo no se observó interacción farmacocinética clínicamente significativa entre la pregabalina y fenitoína, carbamazepina, ácido valproico, lamotrigina, gabapentina, lorazepam, oxicodona o etanol. El análisis farmacocinético poblacional demostró que los medicamentos orales antidiabéticos, diuréticos, insulina, fenobarbital, tiagabina y topiramato no tienen un efecto clínicamente significativo sobre el aclaramiento de la pregabalina.
Anticonceptivos orales, noretisterona y/o etinilestradiol.
La administración concomitante de pregabalina con anticonceptivos orales, noretisterona y/o etinilestradiol no afecta la farmacocinética en estado de equilibrio de ninguno de estos medicamentos.
Medicamentos que afectan al SNC.
La pregabalina puede potenciar los efectos del etanol y del lorazepam. Durante la vigilancia poscomercialización se han notificado casos de insuficiencia respiratoria, coma y muerte en pacientes que tomaban pregabalina junto con opioides y/o otros medicamentos que deprimen la función del SNC. La pregabalina probablemente intensifica los trastornos cognitivos y motores básicos provocados por el oxicodona.
Interacciones en pacientes de edad avanzada.
No se han realizado estudios específicos sobre interacciones farmacodinámicas que incluyan voluntarios de edad avanzada. Los estudios sobre interacciones medicamentosas se han llevado a cabo únicamente en pacientes adultos.
Características de aplicación.
Pacientes con diabetes mellitus.
De acuerdo con la práctica clínica actual, algunos pacientes con diabetes mellitus cuyo peso corporal aumentó durante la terapia con pregabalina pueden requerir ajuste de la dosis de medicamentos hipoglucemiantes.
Reacciones de hipersensibilidad.
Tras la comercialización del medicamento, se han notificado reacciones de hipersensibilidad, incluyendo angioedema. Si aparecen síntomas de angioedema, como hinchazón del rostro, edema perioral o edema de las vías respiratorias superiores, el tratamiento con pregabalina debe interrumpirse inmediatamente.
Reacciones adversas cutáneas graves (RACG)
Durante el tratamiento con pregabalina, rara vez se han notificado reacciones adversas cutáneas graves, incluyendo el síndrome de Stevens-Johnson (SSJ) y la necrólisis epidérmica tóxica (NET), que pueden poner en peligro la vida o ser letales. Al prescribir pregabalina, se debe informar a los pacientes sobre estos signos y síntomas y se debe vigilar cuidadosamente cualquier reacción cutánea. Si aparecen signos o síntomas que sugieran estas reacciones, debe suspenderse inmediatamente la administración de pregabalina y considerarse un tratamiento alternativo (si fuera necesario).
Mareo, somnolencia, pérdida de conciencia, confusión y trastornos psíquicos.
El uso de pregabalina ha estado asociado con mareo y somnolencia, lo que puede aumentar el riesgo de eventos traumáticos (caídas) en pacientes de edad avanzada. Además, tras la comercialización del medicamento, se han notificado casos de pérdida de conciencia, confusión y trastornos psíquicos. Por ello, se debe recomendar a los pacientes que sean precavidos hasta que se conozcan los posibles efectos de este medicamento.
Trastornos visuales.
Durante estudios controlados, la visión borrosa se observó con mayor frecuencia en pacientes que tomaban pregabalina que en aquellos que recibían placebo. En la mayoría de los casos, este fenómeno desapareció al continuar la terapia. En estudios clínicos que incluyeron exámenes oftalmológicos, la frecuencia de casos de deterioro de la agudeza visual y cambios en el campo visual fue mayor en pacientes que recibieron pregabalina en comparación con los del grupo placebo; la frecuencia de cambios en el fondo de ojo fue mayor en pacientes del grupo placebo (véase la sección «Farmacodinámica»).
Tras la comercialización del medicamento, también se han notificado reacciones adversas oculares, incluyendo pérdida de visión, visión borrosa u otros cambios en la agudeza visual, muchos de los cuales fueron temporales. Tras la interrupción del tratamiento con pregabalina, estos síntomas oculares pueden desaparecer o disminuir.
Insuficiencia renal.
Se han notificado casos de insuficiencia renal, que a veces fue reversible tras la interrupción del tratamiento con pregabalina.
Suspensión de medicamentos antiepilépticos concomitantes.
Actualmente hay datos insuficientes sobre si es posible suspender los medicamentos antiepilépticos concomitantes tras alcanzar el control de las convulsiones mediante la adición de pregabalina, con el fin de pasar a monoterapia con pregabalina.
Insuficiencia cardíaca congestiva.
Tras la comercialización del medicamento, se han notificado casos de insuficiencia cardíaca congestiva en algunos pacientes que tomaban pregabalina. Esta reacción se observó principalmente durante el tratamiento con pregabalina para el dolor neuropático en pacientes de edad avanzada con trastornos cardiovasculares preexistentes. En estos pacientes, pregabalina debe administrarse con precaución. Esta reacción puede desaparecer tras la suspensión del tratamiento con pregabalina.
Tratamiento del dolor neuropático de origen central debido a lesión medular.
Durante el tratamiento del dolor neuropático de origen central provocado por lesión medular, aumentó la frecuencia general de reacciones adversas, así como las reacciones adversas del sistema nervioso central, especialmente la somnolencia. Esto puede estar relacionado con un efecto aditivo de los medicamentos concomitantes (por ejemplo, medicamentos antiespásticos) necesarios para tratar esta condición. Esta circunstancia debe tenerse en cuenta al prescribir pregabalina para esta indicación.
Depresión respiratoria.
Se han notificado casos de depresión respiratoria grave asociada con el uso de pregabalina. Los pacientes con función respiratoria alterada, enfermedades respiratorias o neurológicas, insuficiencia renal, uso concomitante de depresores del SNC y personas de edad avanzada pueden tener un mayor riesgo de esta reacción adversa grave. En estos pacientes puede ser necesaria una ajuste de la dosis.
Pensamientos y comportamientos suicidas.
Se han notificado casos de pensamientos y comportamientos suicidas en pacientes que recibían tratamiento con medicamentos antiepilépticos por diversas indicaciones. Según los resultados de un metaanálisis de datos procedentes de estudios aleatorizados y controlados con placebo con medicamentos antiepilépticos, también se observó un ligero aumento del riesgo de aparición de pensamientos y comportamientos suicidas. El mecanismo de este riesgo no se conoce. Tras la comercialización del medicamento, se han notificado casos de pensamientos y comportamientos suicidas en pacientes que recibían pregabalina (véase la sección «Reacciones adversas»). Un estudio epidemiológico utilizando un diseño de control propio (comparación de períodos de tratamiento con períodos sin tratamiento en el mismo paciente) demostró un mayor riesgo de nuevos episodios de comportamiento suicida y muertes por suicidio en pacientes que recibían pregabalina.
A los pacientes (y a las personas que cuidan de ellos) se les debe aconsejar que busquen atención médica si aparecen signos de pensamientos o comportamientos suicidas. Los pacientes deben estar bajo vigilancia para detectar signos de pensamientos o comportamientos suicidas, y se debe considerar el tratamiento adecuado. En caso de pensamientos o comportamientos suicidas, se debe considerar la posibilidad de suspender el tratamiento con pregabalina.
Alteraciones en la función del tracto gastrointestinal inferior.
Tras la comercialización del medicamento, se han notificado eventos relacionados con el deterioro de la función del tracto gastrointestinal inferior (como obstrucción intestinal, obstrucción intestinal paralítica, estreñimiento) durante el uso de pregabalina junto con medicamentos que pueden causar estreñimiento, por ejemplo, analgésicos opioides. Al administrar pregabalina junto con opioides, se deben tomar medidas para prevenir el estreñimiento (especialmente en mujeres y pacientes de edad avanzada).
Uso concomitante con opioides.
Se recomienda precaución al prescribir pregabalina junto con opioides debido al riesgo de depresión del SNC (véase la sección «Interacción con otros medicamentos y otras formas de interacción»). En un estudio de casos y controles en personas que usaban opioides, el riesgo de mortalidad relacionada con opioides fue mayor en pacientes que recibían tratamiento con pregabalina junto con un opioide, en comparación con aquellos que solo usaban opioides (razón de odds ajustada [aOR], 1,68 [IC 95 %, 1,19–2,36]). Este mayor riesgo se observó con dosis bajas de pregabalina (≤ 300 mg, aOR 1,52 [IC 95 %, 1,04–2,22]) y tendía a aumentar con dosis altas de pregabalina (> 300 mg, aOR 2,55 [IC 95 %, 1,24–5,06]).
Uso inadecuado, abuso o dependencia.
La pregabalina puede provocar dependencia medicamentosa, que puede desarrollarse incluso con dosis terapéuticas. Se han notificado casos de abuso y uso inadecuado. Los pacientes con antecedentes de abuso de sustancias psicoactivas pueden tener un mayor riesgo de uso inadecuado, abuso o dependencia de pregabalina, por lo que en estos pacientes debe administrarse con precaución. Antes de prescribir pregabalina, se debe evaluar cuidadosamente el riesgo de uso inadecuado, abuso o dependencia.
Los pacientes que reciben pregabalina deben vigilarse para detectar síntomas de uso inadecuado, abuso o dependencia, como desarrollo de tolerancia, sobredosis o conducta orientada a obtener el medicamento.
Síntomas de retirada.
En algunos pacientes, tras la interrupción de un tratamiento a corto o largo plazo con pregabalina, se han observado síntomas de retirada. Se han notificado síntomas como insomnio, cefalea, náuseas, ansiedad, diarrea, síndrome tipo gripal, inquietud, depresión, dolor, convulsiones, sudoración excesiva y mareo, que indican dependencia medicamentosa. La aparición de síntomas de retirada tras la interrupción de pregabalina puede indicar dependencia medicamentosa (véase la sección «Reacciones adversas»). Esta información debe comunicarse al paciente antes de iniciar el tratamiento.
Si es necesario interrumpir el tratamiento con pregabalina, se recomienda hacerlo gradualmente durante al menos 1 semana, independientemente de la indicación (véase la sección «Posología y forma de administración»).
Las convulsiones, especialmente el estado epiléptico y las convulsiones generalizadas, pueden ocurrir durante el tratamiento con pregabalina o poco después de su suspensión.
Los datos sobre la suspensión de pregabalina tras un uso prolongado indican que la frecuencia y gravedad de los síntomas de retirada pueden depender de la dosis.
Encefalopatía.
Se han notificado casos de encefalopatía, que se presentaron principalmente en pacientes con enfermedades concomitantes que podrían provocar encefalopatía.
Mujeres en edad fértil / anticoncepción.
El uso de pregabalina durante el primer trimestre del embarazo puede provocar malformaciones congénitas importantes en el feto. No debe administrarse pregabalina durante el embarazo a menos que el beneficio para la madre supere claramente el riesgo potencial para el feto. Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento (véase la sección «Uso durante el embarazo o la lactancia»).
Intolerancia a la lactosa.
El medicamento Lyrica contiene lactosa. No debe administrarse a pacientes con enfermedades hereditarias raras como intolerancia a la galactosa, deficiencia de lactasa de Lapp o síndrome de malabsorción de glucosa-galactosa.
Contenido de sodio.
El medicamento Lyrica contiene menos de 1 mmol de sodio (23 mg) por cápsula, es decir, prácticamente libre de sodio. Esta información puede comunicarse a los pacientes que siguen una dieta baja en sodio.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil / métodos anticonceptivos para mujeres.
Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento (véase la sección «Características de aplicación»).
Embarazo.
En estudios en animales se ha demostrado toxicidad reproductiva.
Se ha demostrado que la pregabalina atraviesa la placenta en ratas (véase la sección «Farmacocinética»). La pregabalina puede atravesar la placenta humana.
Malformaciones congénitas importantes.
Según datos de un estudio observacional realizado en países nórdicos, que incluyó más de 2700 embarazos, durante el uso de pregabalina en el primer trimestre se observó una mayor prevalencia de malformaciones congénitas importantes (MCI) en la población infantil (niños vivos o muertos al nacer) expuesta a pregabalina en comparación con la población no expuesta (5,9 % frente a 4,1 %).
El riesgo de desarrollar MCI en niños cuyas madres usaron pregabalina durante el primer trimestre del embarazo fue ligeramente mayor en comparación con niños no expuestos in utero (razón de prevalencia ajustada e IC del 95 %: 1,14 (0,96–1,35)) y en comparación con niños expuestos a lamotrigina (1,29 (1,01–1,65)) o duloxetina (1,39 (1,07–1,82)).
El análisis de malformaciones específicas mostró un mayor riesgo de malformaciones del sistema nervioso, ojos, fisuras orofaciales, vías urinarias y órganos genitales, aunque la cantidad de estos casos fue pequeña y las estimaciones imprecisas.
El medicamento Lyrica no debe usarse durante el embarazo sin una necesidad clara (cuando el beneficio para la madre supere claramente el riesgo potencial para el feto).
Lactancia.
Se ha detectado una pequeña cantidad de pregabalina en la leche materna de mujeres lactantes. Se debe informar a las mujeres lactantes que no se recomienda la lactancia durante el tratamiento con pregabalina.
Fertilidad.
No existen datos clínicos sobre el efecto de la pregabalina en la fertilidad femenina.
En un estudio clínico sobre el efecto de la pregabalina en la movilidad espermática, voluntarios masculinos sanos tomaron pregabalina a una dosis de 600 mg/día. Tras 3 meses de tratamiento, no se observó ningún efecto sobre la movilidad espermática.
En estudios de fertilidad en ratas hembra se observó un efecto adverso sobre la función reproductiva. En estudios de fertilidad en ratas macho se observó un efecto adverso sobre la función reproductiva y el desarrollo. La relevancia clínica de estos resultados es desconocida.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar otros mecanismos.
Lyrica puede tener un efecto leve o moderado sobre la capacidad para conducir vehículos o manejar maquinaria. Lyrica puede causar mareo y somnolencia, y por lo tanto, afectar la capacidad para conducir vehículos o manejar maquinaria. Por ello, se debe recomendar a los pacientes que eviten conducir vehículos, trabajar con maquinaria compleja o realizar otras actividades potencialmente peligrosas hasta que se conozca el efecto de este medicamento sobre su capacidad para realizar dichas actividades.
Vía de administración y dosis.
Vía de administración.
El medicamento Lyrica se toma independientemente de la ingestión de alimentos.
Este medicamento está destinado exclusivamente para administración oral.
Dosis.
El rango de dosis del medicamento puede variar entre 150 y 600 mg por día. La dosis diaria se divide en 2 o 3 tomas.
Dolor neuropático.
El tratamiento con pregabalina puede iniciarse con una dosis de 150 mg por día, dividida en 2 o 3 tomas. Dependiendo de la respuesta individual y la tolerancia del paciente al medicamento, la dosis puede aumentarse hasta 300 mg por día tras 3-7 días, y si es necesario, hasta la dosis máxima de 600 mg por día tras otros 7 días adicionales.
Epilepsia.
El tratamiento con pregabalina puede iniciarse con una dosis de 150 mg por día, dividida en 2 o 3 tomas. Dependiendo de la respuesta individual y la tolerancia del paciente al medicamento, la dosis puede aumentarse hasta 300 mg por día tras la primera semana de tratamiento. Tras otra semana más, la dosis puede aumentarse hasta la máxima de 600 mg por día.
Trastorno de ansiedad generalizada.
La dosis, dividida en 2 o 3 tomas, puede variar entre 150 y 600 mg por día. Debe revisarse periódicamente la necesidad de continuar el tratamiento.
El tratamiento con pregabalina puede iniciarse con una dosis de 150 mg por día. Dependiendo de la respuesta individual y la tolerancia del paciente al medicamento, la dosis puede aumentarse hasta 300 mg por día tras la primera semana de tratamiento. Tras otra semana de toma, la dosis puede aumentarse hasta 450 mg por día. Tras una semana adicional, la dosis puede aumentarse hasta la máxima de 600 mg por día.
Fibromialgia.
La dosis recomendada del medicamento para el tratamiento de la fibromialgia oscila entre 300 y 450 mg por día. El tratamiento debe iniciarse con una dosis de 75 mg dos veces al día (150 mg por día). Dependiendo de la eficacia y la tolerancia, la dosis puede aumentarse hasta 150 mg dos veces al día (300 mg por día) durante la primera semana. En pacientes en los que la dosis de 300 mg por día no sea suficientemente eficaz, puede aumentarse la dosis hasta 225 mg dos veces al día (450 mg por día). Aunque existe un estudio sobre el uso de una dosis de 600 mg por día, no hay evidencia de que esta dosis ofrezca beneficios adicionales; además, esta dosis mostró una peor tolerancia. Debido a las reacciones adversas dependientes de la dosis, no se recomienda el uso de dosis superiores a 450 mg por día. Dado que Lyrica se elimina principalmente por vía renal, debe ajustarse la dosis en pacientes con alteración de la función renal.
Supresión de la pregabalina.
De acuerdo con la práctica clínica actual, se recomienda interrumpir el tratamiento con pregabalina de forma gradual durante al menos una semana, independientemente de la indicación (ver secciones «Precauciones de uso» y «Reacciones adversas»).
Alteración de la función renal.
La pregabalina se elimina del torrente sanguíneo sistémico sin cambios, principalmente por vía renal. Dado que el aclaramiento de la pregabalina es directamente proporcional al aclaramiento de la creatinina (ver sección «Farmacocinética»), la dosis debe ajustarse individualmente en pacientes con alteración de la función renal según se indica en la tabla siguiente, de acuerdo con el aclaramiento de la creatinina (CLcr), que se calcula mediante la fórmula:
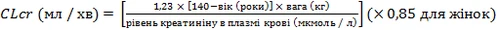
La pregabalina se elimina eficazmente del plasma sanguíneo mediante hemodiálisis (50 % del fármaco en 4 horas). En pacientes sometidos a hemodiálisis, la dosis diaria de pregabalina debe ajustarse según la función renal. Además de la dosis diaria, debe administrarse una dosis adicional inmediatamente después de cada procedimiento de hemodiálisis de 4 horas (ver tabla).
Ajuste de la dosis de pregabalina según la función renal.
| Depuración de creatinina (CLcr) (ml/min) |
Dosis diaria total de pregabalina * |
Esquema de dosificación |
|
| Dosis inicial (mg/día) |
Dosis máxima (mg/día) |
||
| ≥ 60 |
150 |
600 |
Dos o tres veces al día |
| ≥ 30 – < 60 |
75 |
300 |
Dos o tres veces al día |
| ≥ 15 – < 30 |
25–50 |
150 |
Una o dos veces al día |
| < 15 |
25 |
75 |
Una vez al día |
| Dosis adicional tras hemodiálisis (mg) |
|||
| 25 |
100 |
Dosis única+ |
|
* La dosis diaria total (mg/día) debe dividirse en varias tomas según el régimen de dosificación para obtener la dosis por administración única (mg/dosis).
- Dosis adicional: es una dosis única adicional.
Insuficiencia hepática.
No se requiere ajuste de la dosis en pacientes con alteración de la función hepática (ver sección «Farmacocinética»).
Pacientes de edad avanzada.
En pacientes de edad avanzada puede ser necesaria una reducción de la dosis de pregabalina debido a la disminución de la función renal (ver sección «Propiedades farmacodinámicas»).
Niños.
La seguridad y eficacia del medicamento Lyrica en niños (menores de 18 años) no han sido establecidas. La información disponible hasta la fecha se presenta en la sección «Reacciones adversas», así como en las secciones «Farmacodinámica» y «Farmacocinética», sin embargo, basándose en esta información, no es posible formular recomendaciones sobre la dosificación para esta categoría de pacientes.
Sobredosis.
Desde la comercialización del medicamento, se han notificado los efectos adversos más frecuentes tras la sobredosis de pregabalina: somnolencia, confusión, agitación e inquietud. También se han recibido notificaciones sobre convulsiones.
Rara vez se han notificado casos de coma.
El tratamiento de la sobredosis de pregabalina consiste en medidas generales de soporte y, si es necesario, puede incluir hemodiálisis (ver sección «Instrucciones de uso y dosis», tabla).
Reacciones adversas.
En el programa clínico de investigación con pregabalina, más de 8900 pacientes recibieron el medicamento, de los cuales 5600 participaron en estudios doble ciego controlados con placebo. Las reacciones adversas más frecuentes notificadas fueron mareo y somnolencia. Las reacciones adversas generalmente fueron de grado leve o moderado. En todos los estudios controlados, la tasa de interrupción del tratamiento debido a reacciones adversas fue del 12 % en pacientes que recibieron pregabalina y del 5 % en pacientes que recibieron placebo. Las reacciones adversas más frecuentes que condujeron a la interrupción del tratamiento en el grupo de pregabalina fueron mareo y somnolencia.
A continuación se indican todas las reacciones adversas que ocurrieron con mayor frecuencia que con placebo y en más de un paciente; estas reacciones adversas se enumeran por sistemas orgánicos y por frecuencia: muy frecuentes (≥ 1/10); frecuentes (de ≥ 1/100 a < 1/10); poco frecuentes (de ≥ 1/1000 a < 1/100); raras (de ≥ 1/10000 a < 1/1000); muy raras (< 1/10000); frecuencia desconocida (no puede estimarse a partir de los datos disponibles). Dentro de cada grupo por frecuencia, los efectos adversos se presentan en orden descendente de gravedad.
Las reacciones adversas indicadas también pueden estar relacionadas con la evolución de la enfermedad subyacente y/o con el uso concomitante de otros medicamentos.
Durante el tratamiento del dolor neuropático de origen central debido a lesión medular, aumentó la frecuencia general de reacciones adversas, especialmente las del sistema nervioso central (SNC), en particular la somnolencia (ver sección «Precauciones de uso»).
Las reacciones adversas adicionales notificadas tras la comercialización del medicamento se indican a continuación y se destacan en cursiva.
Infecciones e infestaciones.
Frecuentes: nasofaringitis.
Alteraciones de la sangre y del sistema linfático.
Poco frecuentes: neutropenia.
Alteraciones del sistema inmunitario.
Poco frecuentes: hipersensibilidad.
Raras: edema angioneurótico, reacciones alérgicas, reacciones anafilactoides.
Trastornos del metabolismo y de la nutrición.
Frecuentes: aumento del apetito.
Poco frecuentes: pérdida del apetito, hipoglucemia.
Alteraciones del estado psíquico.
Frecuentes: estado eufórico, confusión, irritabilidad, desorientación, insomnio, disminución de la libido.
Poco frecuentes: alucinaciones, ataques de pánico, inquietud, excitación, depresión, estado de ánimo deprimido, estado de ánimo elevado, agresividad, cambios de humor, despersonalización, dificultad para encontrar palabras, sueños patológicos, aumento de la libido, anorgasmia, apatía.
Raras: desinhibición, comportamiento suicida, pensamientos suicidas.
Frecuencia desconocida: dependencia del medicamento.
Alteraciones del sistema nervioso.
Muy frecuentes: mareo, somnolencia, dolor de cabeza.
Frecuentes: ataxia, alteración de la coordinación, temblor, disartria, amnesia, empeoramiento de la memoria, trastorno de la atención, parestesia, hipoestesia, efecto sedante, alteración del equilibrio, letargo.
Poco frecuentes: síncope, estupor, mioclonía, pérdida de conciencia, hiperactividad psicomotriz, discinesia, mareo postural, temblor de intención, nistagmo, alteración de las funciones cognitivas, trastorno mental, trastornos del habla, hiporreflexia, hiperestesia, sensación de ardor, ageusia, malestar general, apatía, parestesia perioral, mioclono.
Raras: convulsiones, parosmia, hipocinesia, disfagia, parkinsonismo, hipalgesia, dependencia, síndrome cerebeloso, síndrome de rueda dentada, coma, delirio, encefalopatía, síndrome extrapiramidal, síndrome de Guillain-Barré, hipertensión intracraneal, reacciones maníacas, reacciones paranoides, trastornos del sueño.
Alteraciones del ojo.
Frecuentes: visión borrosa, diplopía, conjuntivitis.
Poco frecuentes: pérdida de la visión periférica, alteración visual, hinchazón de los ojos, defectos del campo visual, disminución de la agudeza visual, dolor ocular, astenopia, fotopsia, sequedad ocular, lagrimeo excesivo, irritación ocular, blefaritis, alteración de la acomodación, hemorragia ocular, fotofobia, edema de retina.
Raras: pérdida de visión, queratitis, oscilopsia, alteración de la percepción visual de la profundidad, midriasis, estrabismo, brillo visual, anisocoria, úlceras corneales, exoftalmia, parálisis del músculo ocular, iritis, queratoconjuntivitis, miosis, ceguera nocturna, oftalmoplejía, atrofia del nervio óptico, edema de la papila del nervio óptico, ptosis, uveítis.
Alteraciones del oído y del laberinto.
Frecuentes: vértigo.
Poco frecuentes: hiperacusia.
Alteraciones del corazón.
Poco frecuentes: taquicardia, bloqueo auriculoventricular de primer grado, bradicardia sinusal, insuficiencia cardíaca congestiva.
Raras: prolongación del intervalo QT, taquicardia sinusal, arritmia sinusal.
Alteraciones vasculares.
Poco frecuentes: hipotensión arterial, hipertensión arterial, sofocos, hiperemia, sensación de frío en las extremidades.
Alteraciones del aparato respiratorio, del tórax y del mediastino.
Frecuentes: dolor faringolaríngeo.
Poco frecuentes: disnea, epistaxis, tos, congestión nasal, rinitis, ronquidos, sequedad de la mucosa nasal.
Raras: edema pulmonar, sensación de opresión en la garganta, espasmo laríngeo, apnea, atelectasia, bronquiolitis, hipo, fibrosis pulmonar, bostezo.
Frecuencia desconocida: depresión respiratoria.
Alteraciones gastrointestinales.
Frecuentes: vómitos, náuseas, estreñimiento, diarrea, flatulencia, distensión abdominal, sequedad de boca, gastroenteritis.
Poco frecuentes: enfermedad por reflujo gastroesofágico, hipersecreción salival, hipoestesia de la cavidad oral, colecistitis, litiasis biliar, colitis, hemorragia gastrointestinal, melena, edema lingual, hemorragia rectal.
Raras: ascitis, pancreatitis, edema lingual, disfagia, estomatitis aftosa, úlcera esofágica, absceso periodontal.
Alteraciones hepatobiliares.
Poco frecuentes: aumento de las enzimas hepáticas*.
Raras: ictericia.
Muy raras: insuficiencia hepática, hepatitis.
Alteraciones de la piel y del tejido subcutáneo.
Frecuentes: úlceras por presión.
Poco frecuentes: erupción papulosa, urticaria, hiperhidrosis, picor, alopecia, sequedad de la piel, eccema, hirsutismo, úlceras cutáneas, erupción vesiculobullosa.
Raras: necrólisis epidérmica tóxica, síndrome de Stevens-Johnson, sudoración fría, dermatitis exfoliativa, dermatitis liquenoide, melanosis, alteraciones de las uñas, erupción petequial, púrpura, erupción pustulosa, atrofia cutánea, necrosis cutánea, nódulos cutáneos y subcutáneos.
Alteraciones del sistema musculoesquelético y del tejido conjuntivo.
Frecuentes: calambres musculares, artralgia, dolor de espalda, dolor en las extremidades, espasmos musculares del cuello.
Poco frecuentes: hinchazón articular, mialgia, fasciculaciones musculares, dolor de cuello, rigidez muscular.
Raras: rabdomiólisis.
Alteraciones renales y urinarias.
Poco frecuentes: incontinencia urinaria, disuria, albuminuria, hematuria, formación de cálculos renales, nefritis.
Raras: insuficiencia renal, oliguria, retención urinaria, insuficiencia renal aguda, glomerulonefritis, pielonefritis.
Alteraciones del sistema reproductor y de las glándulas mamarias.
Frecuentes: disfunción eréctil, impotencia.
Poco frecuentes: disfunción sexual, retardo de la eyaculación, dismenorrea, dolor de mamas, leucorrea, menorragia, metrorragia.
Raras: amenorrea, secreción mamaria, aumento de las mamas, ginecomastia, cervicitis, balanitis, epididimitis.
Alteraciones generales y en el lugar de administración.
Frecuentes: edema periférico, edema, alteración de la marcha, caídas, sensación de embriaguez, sensaciones inusuales, fatiga aumentada.
Poco frecuentes: edema generalizado, edema facial, rigidez torácica, dolor, calor, sed, escalofríos, debilidad general, malestar, absceso, inflamación del tejido adiposo, reacciones de fotosensibilidad.
Raras: granuloma, automutilación, fibrosis retroperitoneal, shock.
Pruebas de laboratorio.
Frecuentes: aumento de peso.
Poco frecuentes: aumento de la creatinfosfocinasa en sangre, aumento de la glucosa en sangre, disminución del número de plaquetas, aumento de la creatinina en sangre, disminución del potasio en sangre, disminución de peso.
Raras: disminución de leucocitos en sangre.
* Aumento de alaninoaminotransferasa (ALT) y aspartatoaminotransferasa (AST).
En algunos pacientes, tras la interrupción de un tratamiento a corto o largo plazo con pregabalina, se observaron síntomas de abstinencia. Se notificaron síntomas como insomnio, dolor de cabeza, náuseas, ansiedad, diarrea, síndrome tipo gripal, convulsiones, nerviosismo, depresión, dolor, hiperhidrosis y mareo, lo que sugiere dependencia física. Esta información debe comunicarse al paciente antes de iniciar el tratamiento.
Los datos sobre la interrupción de pregabalina tras un uso prolongado indican que la frecuencia y gravedad de los síntomas de abstinencia pueden ser dependientes de la dosis.
Pacientes pediátricos. El perfil de seguridad de pregabalina, establecido en cinco estudios con pacientes pediátricos con crisis epilépticas parciales con o sin generalización secundaria (un estudio de 12 semanas sobre eficacia y seguridad en pacientes de 4 a 16 años, n=295; un estudio de 14 días sobre eficacia y seguridad en pacientes de 1 mes a menos de 4 años, n=175; un estudio de farmacocinética y tolerabilidad, n=65; y dos estudios abiertos de seguridad de 1 año de duración, n=54 y n=431), fue similar al observado en estudios con adultos epilépticos. Las reacciones adversas más frecuentes observadas en el estudio de 12 semanas con pregabalina fueron somnolencia, fiebre, infecciones de las vías respiratorias superiores, aumento del apetito, aumento de peso y nasofaringitis. Las reacciones adversas más frecuentes observadas en el estudio de 14 días con pregabalina fueron somnolencia, infecciones de las vías respiratorias superiores y fiebre (ver secciones «Instrucciones de uso y dosis», «Farmacodinamia» y «Farmacocinética»).
Notificación de reacciones adversas sospechosas. La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite una vigilancia continua de la relación beneficio-riesgo del medicamento.
Período de validez. 3 años.
Condiciones de conservación.
No requiere condiciones especiales de conservación. Mantener en un lugar fuera del alcance de los niños.
Envase.
Para cápsulas de 75 mg y 150 mg: 14 o 21 cápsulas por blíster, 1 o 4 blísteres por caja de cartón.
Para cápsulas de 50 mg y 300 mg: 21 cápsulas por blíster, 1 o 4 blísteres por caja de cartón.
Categoría de dispensación.
Medicamento sujeto a receta médica.
Fabricante.
Pfizer Manufacturing Deutschland GmbH.
Dirección del fabricante y lugar de actividad.
Mooswaldallee 1, 79090 Freiburg im Breisgau, Alemania / Mooswaldallee 1, 79090 Freiburg im Breisgau, Germany.