Emoclot
UcraniaContenido
INSTRUCCIONES DE USO MÉDICO DEL MEDICAMENTO EMOCLAT
Composición:
Principio activo: factor de coagulación sanguínea humana VIII;
1 frasco con polvo contiene 500 UI o 1000 UI de factor de coagulación sanguínea humana VIII;
Excipientes: cloruro de sodio, citrato de sodio, glicina, cloruro de calcio;
el frasco con disolvente contiene 10 ml de agua para preparaciones inyectables.
La solución reconstituida contiene factor de coagulación sanguínea humana VIII a una concentración de 50 UI/ml (500 UI/10 ml) o 100 UI/ml (1000 UI/10 ml).
La actividad específica del medicamento EMOCLAT es aproximadamente de 80 UI/mg de proteína. La actividad (UI) se determina mediante un ensayo cromogénico de acuerdo con la Farmacopea Europea.
El medicamento se fabrica a partir de plasma humano.
El medicamento contiene factor de von Willebrand humano.
Excipientes con efecto conocido: el medicamento contiene hasta 41 mg de sodio por frasco (10 ml).
Forma farmacéutica. Polvo y disolvente para solución para perfusión.
Principales características físico-químicas: polvo: polvo higroscópico blanco o amarillento, o masa frágil; disolvente: líquido incoloro y transparente.
Grupo farmacoterapéutico. Agentes hemostáticos. Factores de coagulación sanguínea VIII. Factor de coagulación VIII. Código ATC B02BD02.
Propiedades farmacológicas.
Farmacodinámica.
El complejo de factor VIII/factor de von Willebrand está compuesto por dos moléculas (factor VIII y factor de von Willebrand) con funciones fisiológicas diferentes.
Tras la administración al paciente con hemofilia, el factor VIII se une al factor de von Willebrand que circula en la sangre del paciente.
El factor VIII activado actúa como cofactor del factor IX activado, acelerando la transformación del factor X en su forma activa. El factor X activado convierte la protrombina en trombina, que a su vez convierte el fibrinógeno en fibrina, permitiendo así la formación del coágulo.
La hemofilia A es un trastorno hereditario ligado al sexo en la coagulación sanguínea, causado por niveles reducidos de factor VIII:C, lo que conduce a hemorragias abundantes en articulaciones, músculos o órganos internos, que ocurren espontáneamente o como resultado de traumatismos accidentales o intervenciones quirúrgicas. La terapia sustitutiva aumenta los niveles plasmáticos del factor VIII y, por tanto, permite corregir temporalmente el déficit del factor y reducir la tendencia a sangrar.
Cabe señalar que la frecuencia anual de hemorragias (FAH) varía con el uso de diferentes concentrados de factor y en diferentes estudios clínicos.
El factor de von Willebrand actúa como estabilizador del factor VIII, media la adhesión de las plaquetas a las zonas de daño vascular y participa en la agregación plaquetaria.
Diez pacientes con hemofilia A grave (mediana de edad 15 años, rango 5–51) con títulos elevados de inhibidores, incluidos en el registro PROFIT gestionado por la Asociación Italiana de Centros de Hemofilia, recibieron tratamiento con el medicamento EMOCLOT para la erradicación de inhibidores mediante terapia de inducción de tolerancia inmune (ITI). Ocho de estos 10 pacientes recibieron ITI de primera línea, y 2 pacientes recibieron tratamiento ITI de urgencia tras un intento previo fallido con otro concentrado de factor VIII. Cinco pacientes recibieron tratamiento diariamente con dosis medias/altas (100/200 UI/kg/día), y otros cinco pacientes recibieron tratamiento con diferentes dosis cada dos días o tres veces por semana (50–150 UI/kg). Una respuesta completa o parcial mantenida, tras un seguimiento medio de 9 años, se obtuvo en el 50 % de los casos. En los 4 pacientes que lograron éxito completo, el período medio de erradicación del inhibidor fue de 26 meses.
Además, en la literatura se ha descrito la experiencia con 11 pacientes (mediana de edad 17 años) con títulos elevados de inhibidores que recibieron terapia ITI con el medicamento EMOCLOT; en total, la ITI fue exitosa en 9 de los 11 pacientes (82 %), con erradicación completa del inhibidor en 4 (36 %) y éxito parcial en 5 (45 %) pacientes.
Niños
125 niños menores de 6 años sin inhibidores, que nunca habían recibido o habían recibido un tratamiento mínimo con factor de coagulación VIII humano, recibieron el medicamento EMOCLOT en el marco de un estudio aleatorizado controlado (SIPPET), cuyo objetivo fue evaluar la frecuencia de aparición de inhibidores en pacientes tratados con factor de coagulación VIII humano derivado del plasma o recombinante. De los 125 pacientes, 61 recibieron EMOCLOT en dosis establecidas para tratamiento a demanda o con fines profilácticos. De estos 61 pacientes, 34 recibieron tratamiento a demanda, 5 pacientes recibieron dosis profilácticas estándar (3 infusiones/semana), 15 pacientes recibieron profilaxis modificada (2 infusiones/semana) y 7 pacientes recibieron diversas combinaciones de regímenes de tratamiento.
Según los resultados del análisis post-hoc, cuyo objetivo fue evaluar la frecuencia anual de hemorragias (FAH) en pacientes que recibieron EMOCLOT, se obtuvieron valores de FAH de 4,2 (342 episodios) en pacientes que siguieron el régimen a demanda, 7,5 (25 episodios) en pacientes que recibieron profilaxis estándar (de los 25 episodios de hemorragia registrados en este grupo, 24 ocurrieron en un solo paciente; al excluir a este paciente del análisis, el valor de FAH disminuyó a 0,24), 5,8 (92 episodios) en pacientes con profilaxis modificada y 5,9 (60 episodios) en pacientes que recibieron diversas combinaciones de regímenes de tratamiento.
Farmacocinética.
Tras la administración del medicamento, aproximadamente 2/3–3/4 del factor VIII permanece en la circulación sanguínea.
El nivel de actividad del factor VIII alcanzado en plasma representa entre el 80 % y el 120 % de la actividad del factor VIII en plasma calculada.
La actividad del factor VIII en plasma disminuye según una curva exponencial bifásica.
En la fase inicial, la distribución entre el compartimento intravascular y otros líquidos corporales ocurre con un período de semivida plasmática de 3–6 horas.
En la siguiente fase más lenta (que probablemente refleja la captación del factor VIII), el período de semivida varía entre 8 y 20 horas, con un promedio de 12 horas, lo que refleja el verdadero período de semivida biológico.
Las propiedades farmacocinéticas del medicamento EMOCLOT fueron estudiadas en el ensayo clínico «Estudio de farmacocinética y eficacia clínica del concentrado Factor VIII EMOCLOT en pacientes con hemofilia A» (código del estudio KV030), realizado con 15 pacientes con hemofilia A grave (con niveles de factor VIII < 1). Los parámetros farmacocinéticos se determinaron tras dos infusiones separadas (dosis de 25 UI/kg), administradas con un intervalo de 3–6 meses. Durante el período entre las dos infusiones, los pacientes recibieron tratamiento con EMOCLOT según su régimen terapéutico habitual (con fines terapéuticos o profilácticos).
Los valores medios de los parámetros farmacocinéticos del medicamento EMOCLOT, determinados durante el estudio, se presentan en la Tabla 1.
Tabla 1
| Indicador |
Primera infusión |
Segunda infusión |
||
| Sin restar los valores iniciales |
Restando los valores iniciales |
Sin restar los valores iniciales |
Restando los valores iniciales |
|
| AUC0-t (MO·ml-1·h) |
10,94 |
9,96 |
10,75 |
8,95 |
| AUC0-∞ (MO·ml-1·h) |
13,08 |
11,22 |
12,07 |
9,89 |
| Cltot (ml·h-1·kg-1) |
2,63 |
2,89 |
2,51 |
2,99 |
| Recuperación progresiva (%) |
2,688 |
2,671 |
||
| t1/2α (h) |
0,543 |
0,768 |
||
| t1/2β (h) |
12,05 |
15,16 |
||
NIños
Aunque no existen datos específicos sobre el uso en niños, escasos datos publicados de estudios de farmacocinética no han demostrado diferencias significativas entre adultos y niños con la misma enfermedad.
Datos preclínicos de seguridad.
El concentrado del factor de coagulación VIII de la sangre humana es un componente natural del plasma humano y actúa de forma similar al factor VIII endógeno.
Los estudios de toxicidad de dosis única no son relevantes, ya que las dosis elevadas provocan hipervolemia.
Los estudios de toxicidad con administración repetida (múltiple) en animales no son posibles debido a la interferencia de los anticuerpos formados contra la proteína heteróloga.
Incluso dosis considerablemente superiores a las recomendadas para el ser humano por kg de peso corporal no han mostrado ningún efecto tóxico en los animales de estudio.
Dado que la experiencia clínica con el uso del factor de coagulación VIII de la sangre humana no ha confirmado su acción carcinógena ni mutagénica, no se considera necesario realizar estudios experimentales, especialmente con especies heterólogas.
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (deficiencia congénita del factor de coagulación sanguínea humana VIII).
Tratamiento del déficit adquirido del factor de coagulación sanguínea humana VIII.
Tratamiento de pacientes con hemofilia que presentan anticuerpos contra el factor de coagulación sanguínea humana VIII (inhibidores: véase también «Precauciones de uso»).
Contraindicaciones.
Hipersensibilidad a la sustancia activa o a cualquiera de los excipientes del medicamento.
Interacción con otros medicamentos y otras formas de interacción.
No se han notificado interacciones entre los factores de coagulación sanguínea humana VIII y otros medicamentos.
Niños
No existen datos específicos en niños.
Características de uso.
Rastreabilidad
Para mejorar la rastreabilidad de medicamentos biológicos, se recomienda registrar el nombre y el número de lote del medicamento EMOCLAT cada vez que se administre a un paciente.
Hipersensibilidad
Puede presentarse hipersensibilidad durante el tratamiento con EMOCLAT.
El medicamento contiene trazas de proteínas humanas distintas del factor VIII. Si aparecen síntomas de hipersensibilidad, se debe aconsejar al paciente que interrumpa inmediatamente la administración del medicamento y que consulte a su médico. Los pacientes deben estar informados sobre los signos precoces de reacciones de hipersensibilidad, incluyendo erupciones cutáneas, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de presentarse un shock, se deben seguir las normas médicas vigentes para el tratamiento del shock.
Información importante sobre los excipientes del medicamento EMOCLAT
Este medicamento contiene hasta 41 mg de sodio por vial (10 ml), lo que representa el 2,05 % de la dosis diaria máxima recomendada de sodio por la OMS para adultos (2 g).
Inhibidores
La formación de anticuerpos neutralizantes (inhibidores) frente al factor de coagulación VIII humano es una complicación conocida en el tratamiento de pacientes con hemofilia A. Estos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, cuya concentración se determina en unidades de Bethesda (UB) por mililitro de plasma mediante un ensayo modificado. El riesgo de aparición de inhibidores está relacionado con la gravedad de la enfermedad y la exposición al factor VIII, siendo más alto durante los primeros 50 días de tratamiento. Aunque este riesgo es infrecuente, persiste durante toda la vida.
La relevancia clínica de la formación de inhibidores depende de su título. Con títulos bajos de inhibidores, el riesgo de respuesta clínica insuficiente es menor que con títulos altos.
En general, se debe realizar un seguimiento cuidadoso de todos los pacientes que reciben tratamiento con factores de coagulación VIII, mediante observación clínica adecuada y pruebas de laboratorio, para detectar la formación de inhibidores. Si al administrar la dosis adecuada no se alcanza el nivel esperado de actividad del factor VIII en plasma o si no se controla la hemorragia con la dosis correspondiente, se debe investigar la presencia de inhibidores frente al factor VIII. En pacientes con niveles altos de inhibidores, la terapia con factor VIII puede resultar ineficaz, por lo que deben considerarse otras opciones terapéuticas. El tratamiento de estos pacientes debe realizarse bajo supervisión médica especializada en el manejo de pacientes con hemofilia e inhibidores frente al factor VIII.
Complicaciones cardiovasculares
En pacientes con factores de riesgo cardiovasculares preexistentes, la terapia sustitutiva con factor VIII puede aumentar dicho riesgo.
Complicaciones relacionadas con el uso de catéteres
Si se requiere acceso venoso central, se debe considerar el riesgo de complicaciones relacionadas con el dispositivo, incluyendo infecciones locales, bacteriemia y trombosis en el sitio de inserción del catéter.
Seguridad viral
Las medidas estándar para prevenir infecciones derivadas del uso de medicamentos obtenidos de sangre o plasma humano incluyen la selección de donantes, la evaluación de lotes individuales y pools de plasma mediante marcadores específicos de infección, así como la implementación de procedimientos eficaces de fabricación para la inactivación/eliminación de virus.
Sin embargo, no puede excluirse por completo la posibilidad de transmisión de agentes infecciosos, incluyendo virus o patógenos desconocidos o emergentes, al administrar medicamentos derivados de sangre o plasma humano.
Se considera que las medidas adoptadas son eficaces frente a virus envueltos, como el virus de la inmunodeficiencia humana (VIH), el virus de la hepatitis B (VHB) y el virus de la hepatitis C (VHC), así como frente a virus no envueltos, como el virus de la hepatitis A (VHA). No obstante, su eficacia puede ser limitada frente a virus no envueltos, como el parvovirus B19. La infección por parvovirus B19 puede ser grave en mujeres embarazadas (infección fetal) y en pacientes con inmunodeficiencia o eritropoyesis aumentada (por ejemplo, anemia hemolítica).
Debe considerarse la conveniencia de vacunaciones adecuadas (contra hepatitis A y B) en pacientes que reciben repetidamente productos de factor de coagulación VIII derivados de plasma humano.
Con el fin de mantener el vínculo entre el paciente y el lote del medicamento, se recomienda encarecidamente registrar el nombre y el número de lote del medicamento EMOCLAT cada vez que se administre.
Pacientes pediátricos
No existen datos específicos en niños.
Uso durante el embarazo o la lactancia
No se han realizado estudios sobre el efecto del factor VIII en la función reproductiva en animales. Debido a la baja incidencia de hemofilia A en mujeres, no hay experiencia clínica en el uso de factor VIII durante el embarazo o la lactancia. Por tanto, el factor VIII debe administrarse durante el embarazo y la lactancia únicamente si las indicaciones son claramente necesarias.
Efecto sobre la capacidad para conducir y utilizar máquinas
EMOCLAT no afecta la capacidad para conducir vehículos ni para manejar maquinaria.
Vía de administración y dosis
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Control del tratamiento
Durante el curso del tratamiento se recomienda realizar determinaciones adecuadas de los niveles del factor VIII para orientar la dosis a administrar y la frecuencia de las administraciones repetidas. La respuesta al factor VIII puede variar en cada paciente individual, mostrando diferentes periodos de semivida y recuperación. Puede ser necesaria una ajuste de la dosis en pacientes con peso corporal insuficiente o excesivo, calculado sobre la base del peso corporal.
En caso de intervenciones quirúrgicas mayores, es especialmente importante garantizar un control preciso de la terapia sustitutiva mediante el análisis de coagulación sanguínea (actividad del factor VIII en el plasma sanguíneo).
Cuando se utiliza un análisis de coagulación in vitro de una sola etapa basado en el tiempo de tromboplastina parcial activada (TCA) para determinar la actividad del factor VIII en muestras de sangre de pacientes, los resultados de la actividad del factor VIII pueden depender significativamente tanto del tipo de reactivo TCA utilizado como del estándar de comparación empleado en el análisis. Asimismo, pueden existir diferencias significativas entre los resultados del análisis obtenidos mediante el método de coagulación de una sola etapa basado en el TCA y mediante el análisis cromogénico según la Farmacopea Europea. Esto es especialmente importante al cambiar de laboratorio y/o de reactivos utilizados para el análisis.
Dosificación
La dosificación y la duración de la terapia sustitutiva dependen del grado de severidad del déficit del factor VIII, la localización e intensidad del sangrado, así como del estado clínico del paciente.
La cantidad de unidades del factor VIII administrada se expresa en unidades internacionales (UI), según el estándar vigente de la OMS para los productos del factor VIII. La actividad del factor VIII en el plasma se expresa en porcentaje (en relación con el plasma normal humano) o en unidades internacionales (en relación con el estándar internacional para el factor VIII en plasma).
La actividad de una unidad internacional (UI) del factor VIII corresponde a la cantidad de factor VIII presente en 1 ml de plasma humano normal.
Tratamiento a demanda
El cálculo de la dosis necesaria de factor VIII se basa en datos empíricos. 1 unidad internacional (UI) de factor VIII por kg de peso corporal aumenta la actividad del factor VIII en plasma entre un 1,5 % y un 2 % de la actividad normal.
La dosis necesaria se determina mediante la siguiente fórmula:
Cantidad necesaria de unidades = peso corporal (kg) × incremento deseado del factor VIII (%) (UI/dl) × 0,4
La cantidad de medicamento a administrar y la frecuencia de administración siempre deben ajustarse a la eficacia clínica en cada caso particular.
En los siguientes casos de sangrado, la actividad del factor VIII no debe ser inferior al nivel indicado de actividad en plasma sanguíneo (en % respecto a la normalidad) durante el período correspondiente. La Tabla 2 puede utilizarse como guía para la dosificación en casos de sangrado y procedimientos quirúrgicos.
Tabla 2
| Grado de hemorragia / tipo de procedimiento quirúrgico |
Nivel necesario del factor VIII (%) (UI/dl) |
Frecuencia de administración (horas) / duración del tratamiento (días) |
| Hemorragia |
||
| Hemartrosis precoz, hemorragia muscular o hemorragia en la cavidad oral |
20–40 |
Repetir cada 12–24 horas durante al menos 1 día hasta la cesación de la hemorragia, evidenciada por la desaparición del dolor o la cicatrización. |
| Hemartrosis más pronunciada, hemorragia muscular o hematoma |
30–60 |
Repetir la administración cada 12–24 horas durante 3–4 días o más, hasta que desaparezcan el dolor y la discapacidad aguda. |
| Hemorragia amenazante para la vida |
60–100 |
Repetir la administración cada 8–24 horas hasta que desaparezca el riesgo vital. |
| Intervenciones quirúrgicas |
||
| Pequeñas intervenciones quirúrgicas, incluida la extracción dental |
30–60 |
Cada 24 horas durante al menos 1 día hasta la cicatrización. |
| Grandes intervenciones quirúrgicas |
80–100 (antes y después de la intervención quirúrgica) |
Repetir la administración cada 8–24 horas hasta una adecuada cicatrización de la herida; posteriormente continuar el tratamiento durante al menos 7 días, manteniendo la actividad del factor VIII entre el 30–60 % (30–60 UI/dl). |
Prevención
Para la prevención prolongada de hemorragias, a los pacientes con hemofilia A grave se les suelen administrar dosis de 20 a 40 UI del factor VIII por kg de peso corporal, con intervalos de administración de 2 a 3 días. En casos individuales, especialmente en pacientes jóvenes, pueden ser necesarios intervalos más cortos entre las administraciones o dosis más altas.
Vía de administración
Se administra por vía intravenosa mediante inyección o infusión lenta.
En el caso de inyección intravenosa, se recomienda administrar el medicamento durante 3-5 minutos, controlando simultáneamente la frecuencia del pulso del paciente y deteniendo o reduciendo la velocidad de administración si se produce un aumento de la frecuencia cardíaca.
La velocidad de infusión debe determinarse individualmente para cada paciente.
Reconstitución del polvo con el disolvente
- Llevar el frasco con polvo y el frasco con disolvente a temperatura ambiente.
- Mantener la temperatura ambiente durante todo el proceso de reconstitución (disolución) (máximo 10 minutos).
- Retirar las tapas protectoras del frasco con polvo y del frasco con disolvente.
- Desinfectar con alcohol etílico las superficies de los tapones de ambos frascos.
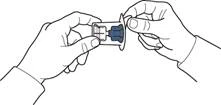
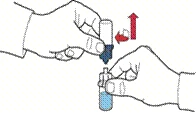
- Abrir el envase del dispositivo según se indica en la figura A, sin tocar la parte interna del envase (figura A).
- No extraer el dispositivo del envase.
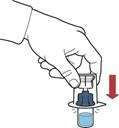
- Dar la vuelta al envase con el dispositivo y perforar con la espiga de plástico del dispositivo el tapón del frasco con disolvente, de modo que la parte azul del dispositivo quede conectada al frasco con disolvente (figura B).
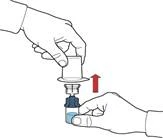
- Retirar el envase del dispositivo sujetándolo por el borde, sin tocar el dispositivo en sí (figura C).
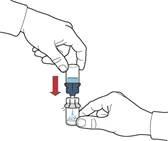
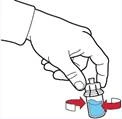
- Asegurarse de que el frasco con polvo esté colocado sobre una superficie segura; dar la vuelta al frasco con disolvente conectado al dispositivo, de forma que el frasco con disolvente quede por encima del dispositivo; presionar el adaptador transparente sobre el tapón del frasco con polvo para que la espiga de plástico atraviese el tapón del frasco con polvo; el disolvente pasará automáticamente al frasco con polvo (figura D).
- Tras la transferencia del disolvente, desenroscar la parte azul del sistema, a la que va unido el frasco con disolvente, y retirarla (figura E).
- Agitar suavemente el frasco hasta que el polvo se disuelva completamente (figura F).
- No agitar vigorosamente el frasco para evitar la formación de espuma.
| Fig. A |
Fig. B |
|
|
|
| Fig. B |
Fig. C |
|
|
|
| Fig. D |
Fig. E |
|
|
|
Administración de la solución
Después de la reconstitución, la solución puede contener una pequeña cantidad de hilos finos o partículas.
Antes de la administración, se debe examinar visualmente la solución para detectar la presencia de partículas o cambios de color. La solución debe ser transparente o ligeramente opalescente. No utilice la solución si está turbia o si contiene un sedimento.
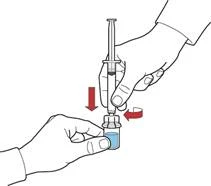
- Introduzca aire en la jeringa tirando del émbolo; conecte la jeringa al dispositivo e introduzca el aire de la jeringa en el frasco que contiene la solución reconstituida (fig. E).
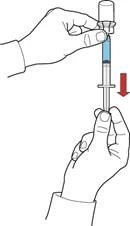
- Manteniendo el émbolo, invierta el sistema de forma que el frasco con la solución reconstituida quede por encima del dispositivo. Tirando lentamente del émbolo, aspire el concentrado a la jeringa (fig. F).
- Desconecte la jeringa girándola en sentido antihorario.
- Examine visualmente la solución en la jeringa, que debe ser transparente o ligeramente opalescente y no debe contener partículas.
- Conecte una aguja-borla a la jeringa y administre el medicamento por vía intravenosa mediante infusión o inyección lenta.
| Fig. E |
Fig. F |
|
|
|
Después de abrirlo, el contenido del frasco debe utilizarse inmediatamente.
La solución reconstituida, una vez cargada en la jeringa, debe utilizarse inmediatamente.
El contenido del frasco debe utilizarse únicamente para una sola administración.
Está prohibido el uso del medicamento después de la fecha de caducidad indicada en el envase.
Cualquier medicamento no utilizado o residuos derivados de su uso deben eliminarse de acuerdo con los requisitos locales.
Niños.
La seguridad y eficacia del uso de EMOCLOT en niños menores de 12 años no han sido establecidas. Los datos disponibles se describen en la sección «Farmacodinamia», pero no pueden darse recomendaciones sobre la dosificación.
La dosificación en adolescentes (12–18 años) para cada indicación se calcula según el peso corporal.
Sobredosificación.
No se han notificado síntomas de sobredosificación con el factor de coagulación VIII humano.
Reacciones adversas.
Descripción breve del perfil de seguridad
Se han observado sensibilidad aumentada o reacciones alérgicas (incluyendo angioedema, dolor ardiente y agudo en el sitio de infusión, escalofríos, sofocos, urticaria generalizada, cefalea, erupción cutánea, hipotensión, letargia, náuseas, inquietud, taquicardia, opresión en el pecho, hormigueo, vómitos y sibilancias) de forma rara, y en algunos casos pueden progresar hasta una anafilaxia grave (incluyendo shock).
También se ha observado aumento de la temperatura.
En pacientes con hemofilia A que reciben tratamiento con factores VIII, incluyendo EMOKLOT, pueden desarrollarse anticuerpos neutralizantes (inhibidores). La aparición de inhibidores en pacientes con hemofilia A se manifestará como una respuesta clínica insuficiente. En tales casos, se recomienda consultar a un centro especializado en hemofilia.
Para información sobre la seguridad respecto a agentes transmisibles, véase la sección «Instrucciones de uso».
Lista de reacciones adversas en forma de tabla
Las reacciones adversas que pueden ocurrir con la administración del factor de coagulación VIII humano se presentan en la Tabla 3, clasificadas según el sistema de órganos del MedDRA [Diccionario Médico para Actividades Regulatorias] y los términos de preferencia.
La frecuencia se ha clasificado según las siguientes categorías convencionales: muy frecuente (≥ 1/10); frecuente (≥ 1/100 a < 1/10); poco frecuente (≥ 1/1 000 a < 1/100); raro (≥ 1/10 000 a < 1/1 000); muy raro (< 1/10 000); frecuencia desconocida (no puede estimarse con los datos disponibles).
No existen datos confirmados sobre la frecuencia de reacciones adversas en estudios clínicos.
Los datos siguientes se basan en el perfil de seguridad del factor de coagulación VIII humano y se han observado parcialmente durante el período poscomercialización del medicamento (experiencia poscomercialización); dado que los informes de reacciones adversas poscomercialización se realizan de forma voluntaria y se refieren a una población de tamaño desconocido, no es posible estimar la frecuencia de dichas reacciones.
Tabla 3
| Clase de sistema de órganos según MedDRA |
Reacción adversa |
Frecuencia |
| Trastornos de la sangre y del sistema linfático |
Aparición de inhibidores del factor VIII |
Infrecuente (PRL) ** Muy frecuente (PRN) ** |
| Trastornos del sistema inmunitario |
Hipersensibilidad |
No conocida |
| Reacciones alérgicas (hipersensibilidad)* |
No conocida |
|
| Reacción anafiláctica |
No conocida |
|
| Shock anafiláctico |
No conocida |
|
| Trastornos psiquiátricos |
Inquietud |
No conocida |
| Trastornos del sistema nervioso |
Cefalea |
No conocida |
| Letergia |
No conocida |
|
| Parastesia |
No conocida |
|
| Trastornos cardíacos |
Taquicardia |
No conocida |
| Trastornos vasculares |
Palpitaciones |
No conocida |
| Hipotensión |
No conocida |
|
| Trastornos respiratorios, torácicos y mediastínicos |
Disnea sibilante* |
No conocida |
| Trastornos gastrointestinales |
Náuseas |
No conocida |
| Vómitos |
No conocida |
|
| Trastornos de la piel y del tejido subcutáneo |
Angioedema |
No conocida |
| Urticaria generalizada (urticaria)* |
No conocida |
|
| Erupción cutánea (urticaria)* |
No conocida |
|
| Trastornos generales y condiciones en el sitio de administración |
Dolor ardoroso en el sitio de infusión (dolor en el sitio de infusión)* |
No conocida |
| Dolor agudo en el sitio de infusión (dolor en el sitio de infusión)* |
No conocida |
|
| Escalofríos |
No conocida |
|
| Opresión en el pecho (malestar en el pecho)* |
No conocida |
|
| Pirexia |
No conocida |
* Términos MedDRA de nivel inferior que describen mejor estas reacciones adversas; entre paréntesis se indica el término MedDRA de uso preferente.
** Los datos sobre frecuencia se basan en los resultados de estudios clínicos con todos los medicamentos del factor VIII, realizados en pacientes con hemofilia A grave. PTL: pacientes previamente tratados; PTN: pacientes previamente no tratados.
Niños
No existen datos específicos en niños.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es importante. Permite una vigilancia continua de la relación beneficio/riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar sobre todos los casos de reacciones adversas sospechosas y sobre la falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en el enlace: https://aisf.dec.gov.ua.
Periodo de validez.
3 años.
Diluyente (agua para inyecciones): 5 años.
El medicamento debe usarse inmediatamente después de la reconstitución.
Condiciones de conservación.
Conservar en nevera (entre 2 y 8 ℃). No congelar. Conservar en el embalaje original para protegerlo de la luz, en un lugar fuera del alcance de los niños.
Antes de la administración y durante su periodo de validez, el frasco con polvo puede conservarse a temperatura ambiente no superior a 25 ℃ durante un período continuo de hasta 6 meses. Tras este período, el frasco con polvo debe destruirse.
Después del almacenamiento del frasco con polvo a temperatura ambiente, no se permite su almacenamiento posterior en nevera.
La fecha de inicio del almacenamiento del medicamento a temperatura ambiente debe anotarse en el embalaje original del medicamento, en el espacio especialmente previsto para ello.
Incompatibilidades.
Debido a la falta de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Debe utilizarse únicamente el conjunto para inyección/infusión suministrado, ya que el tratamiento podría no ser eficaz debido a la adsorción del factor VIII de coagulación sanguínea humana en las superficies internas de otros dispositivos de administración.
Envase.
500 UI o 1000 UI por frasco Nº 1, acompañado de diluyente (agua para inyecciones) 10 ml por frasco Nº 1 y un conjunto para reconstitución y administración, en caja de cartón.
Categoría de dispensación. Bajo receta.
Fabricante.
KEDRION S.P.A.
Domicilio del fabricante y dirección del lugar de actividad.
VIA PROVINCIALE (loc. BOLOGNANA) - 55027 GALLICANO (LU), Italia.