Emoclot
UkraineTable of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT EMOCLAT
Composition:
Active substance: human blood coagulation factor VIII;
1 vial of powder contains 500 IU or 1000 IU of human blood coagulation factor VIII;
Excipients: sodium chloride, sodium citrate, glycine, calcium chloride;
the vial with solvent contains 10 ml of water for injections.
The reconstituted solution contains human blood coagulation factor VIII at a concentration of 50 IU/ml (500 IU/10 ml) or 100 IU/ml (1000 IU/10 ml).
The specific activity of EMOCLAT is approximately 80 IU/mg protein. The activity (IU) is determined by chromogenic assay according to the European Pharmacopoeia.
The product is manufactured from human plasma.
The product contains human von Willebrand factor.
Excipients with known effect: the medicinal product contains up to 41 mg of sodium per vial (10 ml).
Pharmaceutical form. Powder and solvent for solution for infusion.
Main physicochemical characteristics: powder: white or pale yellow hygroscopic powder or friable mass; solvent: clear, colorless liquid.
Pharmacotherapeutic group. Haemostatics. Coagulation factors VIII. Coagulation factor VIII. ATC code B02BD02.
Pharmacological Properties.
Pharmacodynamics.
The factor VIII/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions.
When administered to a patient with hemophilia, factor VIII binds to the von Willebrand factor circulating in the patient's blood.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X into its active form. Activated factor X converts prothrombin into thrombin, which in turn converts fibrinogen into fibrin, allowing clot formation.
Hemophilia A is an X-linked inherited coagulation disorder caused by reduced levels of factor VIII:C, leading to severe bleeding episodes into joints, muscles, or internal organs, occurring spontaneously or following trauma or surgical procedures. Replacement therapy increases plasma levels of factor VIII and thus temporarily corrects the deficiency, reducing the tendency to bleed.
It should be noted that the annualized bleeding rate (ABR) varies with different factor concentrates and across different clinical studies.
Von Willebrand factor acts as a stabilizer of factor VIII, mediates platelet adhesion to sites of vascular injury, and participates in platelet aggregation.
Ten patients with severe hemophilia A (median age 15 years, range 5–51) with high-titer inhibitors, enrolled in the PROFIT registry managed by the Italian Association of Hemophilia Centers, received EMOCLOT for inhibitor eradication through immune tolerance induction (ITI) therapy. Eight of these 10 patients received first-line ITI, while two underwent emergency ITI therapy after a previous failed attempt using another factor VIII concentrate. Five patients received daily treatment with medium/high doses (100/200 IU/kg/day), and five patients received various alternate-day or three-times-weekly regimens (50–150 IU/kg). Complete or partial sustained response after a median follow-up of 9 years was achieved in 50% of cases. Among the four patients who achieved complete success, the median time to inhibitor eradication was 26 months.
Additionally, the literature describes experience with 11 patients (median age 17 years) with high-titer inhibitors who underwent ITI therapy with EMOCLOT; overall, ITI was successful in 9 out of 11 patients (82%), with complete inhibitor eradication in 4 (36%) and partial success in 5 (45%) patients.
Children
One hundred twenty-five children under 6 years of age without inhibitors, who were previously untreated or minimally treated with human coagulation factor VIII, received EMOCLOT in a controlled randomized trial (SIPPET), designed to evaluate the incidence of inhibitor development in patients treated with plasma-derived or recombinant human coagulation factor VIII. Sixty-one out of 125 patients received EMOCLOT at doses established for on-demand treatment or prophylaxis. Of the 61 patients, 34 received on-demand treatment, 5 received standard prophylaxis (3 infusions/week), 15 received modified prophylaxis (2 infusions/week), and 7 received various combinations of treatment regimens.
According to post-hoc analysis evaluating the annualized bleeding rate (ABR) in patients receiving EMOCLOT, the ABR was 4.2 (342 episodes) in patients on on-demand treatment, 7.5 (25 episodes) in patients on standard prophylaxis (of the total 25 bleeding episodes recorded in this group, 24 occurred in one patient; excluding this patient, the ABR decreased to 0.24), 5.8 (92 episodes) in patients on modified prophylaxis, and 5.9 (60 episodes) in patients receiving various combinations of treatment regimens.
Pharmacokinetics.
After administration, approximately 2/3–3/4 of factor VIII remains in the circulating blood.
The plasma factor VIII activity achieved is 80–120% of the calculated plasma factor VIII activity.
Factor VIII activity in plasma decreases according to a biphasic exponential curve.
In the initial phase, distribution between intravascular and other body fluids occurs with a plasma half-life of 3–6 hours.
In the subsequent slower phase (likely reflecting factor VIII uptake), the half-life ranges from 8 to 20 hours, averaging 12 hours, reflecting the true biological half-life.
The pharmacokinetic properties of EMOCLOT were studied in the clinical trial "Study of Pharmacokinetics and Clinical Efficacy of Factor VIII Concentrate EMOCLOT in Patients with Hemophilia A" (study code KB030), involving 15 patients with severe hemophilia A (with factor VIII levels <1%). Pharmacokinetic parameters were determined after two separate infusions (dose 25 IU/kg) administered 3–6 months apart. Between the two infusions, patients continued receiving EMOCLOT according to their usual therapeutic regimen (for treatment or prophylaxis).
The mean pharmacokinetic parameters of EMOCLOT observed during the study are presented in Table 1.
Table 1
| Parameter |
First infusion |
Second infusion |
||
| Without subtraction of baseline values |
With subtraction of baseline values |
Without subtraction of baseline values |
With subtraction of baseline values |
|
| AUC0-t (MO·ml-1·h) |
10.94 |
9.96 |
10.75 |
8.95 |
| AUC0-∞ (MO·ml-1·h) |
13.08 |
11.22 |
12.07 |
9.89 |
| Cltot (ml·h-1·kg-1) |
2.63 |
2.89 |
2.51 |
2.99 |
| Gradual recovery (%) |
2.688 |
2.671 |
||
| t1/2α (h) |
0.543 |
0.768 |
||
| t1/2β (h) |
12.05 |
15.16 |
||
Children
Although specific pediatric data are lacking, limited published pharmacokinetic studies have not demonstrated significant differences between adults and children with the same condition.
Preclinical safety data.
Human coagulation factor VIII concentrate is a natural component of human plasma and acts similarly to endogenous factor VIII.
Single-dose toxicity studies are not relevant, as high doses cause hypervolemia.
Repeated-dose (multiple-dose) toxicity studies in animals are not feasible due to interference from antibodies formed against the heterologous protein.
Even doses substantially exceeding the recommended human dose per kg of body weight show no toxic effects in experimental animals.
Given the clinical experience with human coagulation factor VIII, in which no carcinogenic or mutagenic effects have been demonstrated, experimental studies, particularly those involving heterologous species, are not considered necessary.
Clinical characteristics.
Indications.
Treatment and prevention of bleeding in patients with hemophilia A (congenital deficiency of human blood coagulation factor VIII).
Treatment of acquired deficiency of human blood coagulation factor VIII.
Treatment of hemophilia patients with antibodies to human blood coagulation factor VIII (inhibitors: see also "Special precautions").
Contraindications.
Hypersensitivity to the active substance or to any of the excipients of the medicinal product.
Interaction with other medicinal products and other forms of interaction.
No interactions between factor VIII coagulation products and other medicinal products have been reported.
Children
Specific data in children are lacking.
Special precautions for use.
Traceability
To improve traceability of biological medicinal products, it is recommended that the name and batch number of the medicinal product EMOCLAT be recorded whenever the product is administered to a patient.
Hypersensitivity
Hypersensitivity reactions may occur during treatment with EMOCLAT.
The product contains traces of human proteins other than factor VIII. If symptoms of hypersensitivity occur, patients should be advised to discontinue the use of the product immediately and contact their physician. Patients should be informed about early signs of hypersensitivity reactions, including rash, generalized urticaria, tightness of the chest, wheezing, hypotension, and anaphylaxis.
If shock occurs, current medical standards for the treatment of shock should be followed.
Important information regarding excipients of the medicinal product EMOCLAT
This medicinal product contains up to 41 mg of sodium per vial (10 mL), which corresponds to 2.05% of the WHO recommended maximum daily intake of sodium for adults (2 g).
Inhibitors
The development of neutralizing antibodies (inhibitors) to human blood coagulation factor VIII is a known complication in the treatment of patients with hemophilia A. These inhibitors are usually immunoglobulins of the IgG class directed against the procoagulant activity of factor VIII. Their concentration is measured in Bethesda Units (BU) per 1 mL of plasma using a modified assay. The risk of inhibitor development correlates with the severity of the disease and exposure to factor VIII, and is highest during the first 50 days of treatment. Although this risk is infrequent, it persists throughout life.
The clinical significance of inhibitor formation depends on their titer. With low inhibitor titers, the risk of inadequate clinical response is lower than with high inhibitor titers.
Overall, all patients receiving treatment with factor VIII coagulation factor products should be carefully monitored for the development of inhibitors through appropriate clinical observation and laboratory testing. If the expected plasma factor VIII activity level is not achieved after administration of an appropriate dose, or if bleeding is not adequately controlled with an appropriate dose, testing for the presence of factor VIII inhibitors should be performed. In patients with high levels of inhibitors, factor VIII therapy may be ineffective, and alternative treatment options should be considered. The treatment of such patients should be managed by a physician experienced in the treatment of patients with hemophilia and factor VIII inhibitors.
Cardiovascular complications
In patients with existing risk factors for cardiovascular disease, replacement therapy with factor VIII may increase this risk.
Complications related to catheter use
If central venous access is required, the risk of device-related complications, including local infections, bacteremia, and thrombosis at the catheter site, should be considered.
Viral safety
Standard precautions to prevent infections from medicinal products derived from human blood or plasma include donor selection, testing of individual plasma pools for specific infectious markers, and effective manufacturing steps for virus inactivation/removal.
Nevertheless, when administering medicinal products manufactured from human blood or plasma, the possibility of transmitting infectious agents, including unknown or emerging viruses and other pathogens, cannot be completely excluded.
The measures taken are considered effective against enveloped viruses such as human immunodeficiency virus (HIV), hepatitis B virus (HBV), and hepatitis C virus (HCV), as well as against non-enveloped viruses such as hepatitis A virus (HAV). However, the measures may have limited effectiveness against non-enveloped viruses such as parvovirus B19. Parvovirus B19 infection may be serious in pregnant women (fetal infection) and in patients with immunodeficiency or increased erythropoiesis (e.g., in hemolytic anemia).
Appropriate vaccination (against hepatitis A and B) should be considered for patients who are regularly or repeatedly treated with plasma-derived factor VIII coagulation factor products.
To maintain the link between the patient and the product batch, it is strongly recommended to record the name and batch number of EMOCLAT each time the product is administered.
Children
Specific data in pediatric patients are lacking.
Use during pregnancy or breastfeeding
Animal studies on the effect of factor VIII on reproductive function have not been conducted. Since the incidence of hemophilia A in women is low, experience with the use of factor VIII during pregnancy and breastfeeding is lacking. Therefore, factor VIII should be used during pregnancy and breastfeeding only if clearly indicated.
Ability to affect reaction speed when driving or operating machinery
EMOCLAT does not affect the ability to drive vehicles or operate machinery.
Dosage and Administration
Treatment should be initiated under the supervision of a physician experienced in the management of hemophilia.
Monitoring of Treatment
During the course of treatment, appropriate monitoring of factor VIII levels is recommended to guide dosing and frequency of repeat administrations. Individual patients may respond differently to factor VIII, demonstrating varying half-lives and recovery times. Dose adjustments may be necessary for patients with body weight that is significantly below or above normal, based on body weight calculations.
In the case of major surgical procedures, it is essential to ensure precise monitoring of replacement therapy by performing coagulation tests (factor VIII activity in plasma).
When using a one-stage in vitro coagulation assay based on activated partial thromboplastin time (aPTT) to determine factor VIII activity in patient plasma samples, results may vary significantly depending on both the type of aPTT reagent and the reference standard used. Considerable discrepancies may also occur between results obtained using the one-stage aPTT-based coagulation assay and chromogenic assays according to the European Pharmacopoeia. This is particularly important when changing laboratories and/or reagents used for testing.
Dosage
The dosage and duration of replacement therapy depend on the severity of factor VIII deficiency, the location and intensity of bleeding, and the patient’s clinical condition.
The administered quantity of factor VIII is expressed in International Units (IU), corresponding to the current World Health Organization (WHO) standard for factor VIII preparations. Factor VIII activity in plasma is expressed as a percentage (relative to normal human plasma) or in International Units (relative to the International Standard for factor VIII in plasma).
One International Unit (IU) of factor VIII activity corresponds to the amount of factor VIII present in 1 ml of normal human plasma.
On-Demand Treatment
The calculation of the required factor VIII dose is based on empirical data. 1 International Unit (IU) of factor VIII per 1 kg of body weight raises plasma factor VIII activity by 1.5–2% of normal activity.
The required dose can be calculated using the following formula:
Required number of units = body weight (kg) × desired increase in factor VIII activity (%) (IU/dL) × 0.4
The amount of product to be administered and the frequency of administration should always be adjusted according to the clinical response in each individual case.
In the situations listed below, factor VIII activity should not fall below the specified plasma activity level (% of normal) during the indicated period. Table 2 can be used as a guideline for dosing in bleeding episodes and surgical procedures.
Table 2
| Degree of bleeding / type of surgical procedure |
Required factor VIII level (%) (IU/dl) |
Dosing frequency (hours) / duration of treatment (days) |
| Bleeding |
||
| Early haemarthrosis, muscle bleeding, or oral cavity bleeding |
20–40 |
Repeat every 12–24 hours for at least 1 day until bleeding stops, as indicated by relief of pain or healing. |
| More pronounced haemarthrosis, muscle bleeding, or hematoma |
30–60 |
Repeat every 12–24 hours for 3–4 days or longer, until pain and acute disability have resolved. |
| Life-threatening bleeding |
60–100 |
Repeat every 8–24 hours until the life-threatening condition has resolved. |
| Surgical procedures |
||
| Minor surgical procedures, including tooth extraction |
30–60 |
Every 24 hours for at least 1 day until healing is achieved. |
| Major surgical procedures |
80–100 (before and after surgery) |
Repeat every 8–24 hours until adequate wound healing; then continue therapy for at least 7 days, maintaining factor VIII activity at 30–60% (30–60 IU/dl). |
Prevention
For long-term prevention of bleeding episodes, patients with severe hemophilia A are usually administered doses of 20 to 40 IU of factor VIII per kg of body weight every 2 to 3 days. In individual cases, especially in younger patients, shorter intervals between doses or higher doses may be required.
Method of administration
The drug is administered intravenously by injection or slow infusion.
When administered by intravenous injection, the product should be injected over 3–5 minutes, with monitoring of the patient's pulse rate. Infusion should be interrupted or the rate reduced if an increase in pulse rate occurs.
The infusion rate should be individually determined for each patient.
Reconstitution of the powder with solvent
- Allow the vial of powder and the vial of solvent to reach room temperature.
- Maintain room temperature throughout the entire reconstitution (dissolving) process (maximum 10 minutes).
- Remove the protective caps from both the vial of powder and the vial of solvent.
- Disinfect the stopper surfaces of both vials with ethyl alcohol.
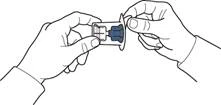
- Open the device packaging as shown in Figure A, without touching the inner part of the packaging (Figure A).
- Do not remove the device from the packaging.
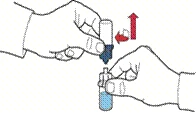
- Turn the packaging with the device and pierce the solvent vial stopper with the plastic spike of the device, so that the blue part of the device connects to the solvent vial (Figure B).
- Remove the device packaging by holding its edge, without touching the device itself (Figure V).
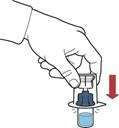
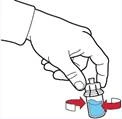
- Ensure the powder vial is placed on a stable surface; invert the solvent vial with the attached device so that the solvent vial is above the device; press the transparent adapter on the powder vial stopper to allow the plastic spike to pierce the powder vial stopper; the solvent will automatically begin to transfer into the powder vial (Figure G).
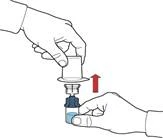
- After the solvent transfer, unscrew the blue part of the system, to which the solvent vial is attached, and remove it (Figure D).
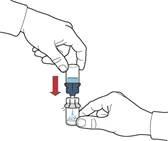
- Gently swirl the vial until the powder is completely dissolved (Figure E).
- Do not shake the vial vigorously to avoid foaming.
| Fig. A |
Fig. B |
|
|
|
| Fig. B |
Fig. C |
|
|
|
| Fig. D |
Fig. E |
|
|
|
Administration of the solution
After reconstitution, the solution may contain a small amount of fine threads or particles.
The solution should be visually inspected for particles or discoloration prior to administration. The solution should be clear or slightly opalescent. Do not use the solution if it is cloudy or contains precipitate.
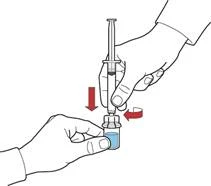
- Draw air into the syringe by pulling back the plunger; attach the syringe to the device and inject the air from the syringe into the vial containing the reconstituted solution (Fig. E).
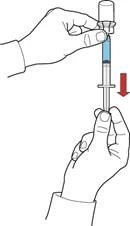
- While holding the plunger, invert the system so that the vial with the reconstituted solution is positioned above the device. Slowly pull back the plunger to draw the concentrate into the syringe (Fig. F).
- Detach the syringe by rotating it counterclockwise.
- Visually inspect the solution in the syringe, which should be clear or slightly opalescent and must not contain particles.
- Attach a butterfly needle to the syringe and administer the medication intravenously by infusion or slow injection.
| Fig. E |
Fig. F |
|
|
|
After opening, the contents of the vial should be used immediately.
The reconstituted solution filled into a syringe should be used immediately.
The contents of the vial should be used for a single administration only.
Use of the medicinal product after the expiry date stated on the packaging is prohibited.
Any unused medicinal product or waste material resulting from its use should be disposed of in accordance with local requirements.
Children.
The safety and efficacy of EMOCLOT in children under 12 years of age have not been established. Available data are described in section "Pharmacodynamics", but dosage recommendations cannot be provided.
Dosage for adolescents (12–18 years) for each indication is calculated based on body weight.
Overdose.
There have been no reports of symptoms related to overdose with human blood coagulation factor VIII.
Adverse reactions.
Summary of safety profile
Hypersensitivity or allergic reactions (including angioneurotic edema, burning and stinging pain at the infusion site, chills, flushing, generalized urticaria, headache, rash, hypotension, lethargy, nausea, restlessness, tachycardia, chest tightness, tingling, vomiting, and wheezing) are rare and in some cases may progress to severe anaphylaxis (including shock).
Elevated body temperature has also been observed.
In patients with hemophilia A receiving treatment with factor VIII products, including EMOKLOT, neutralizing antibodies (inhibitors) may develop. The appearance of inhibitors in patients with hemophilia A is manifested by suboptimal clinical response. In such cases, it is recommended to consult a specialized hemophilia center.
Information on safety regarding transmissible agents is provided in the section "Special precautions for use".
List of adverse reactions in tabular form
Adverse effects that may occur during administration of human blood coagulation factor VIII are presented in Table 3 according to MedDRA system organ class [Medical Dictionary for Regulatory Activities] and preferred terms.
Frequency was estimated using the following categories: very common (≥ 1/10); common (≥ 1/100 to < 1/10); uncommon (≥ 1/1,000 to < 1/100); rare (≥ 1/10,000 to < 1/1,000); very rare (< 1/10,000); frequency not known (cannot be estimated from the available data).
Confirmed data on the frequency of adverse reactions from clinical trials are lacking.
The information listed below is based on the safety profile of human blood coagulation factor VIII and has been partially observed during the post-marketing period (post-marketing experience); since post-marketing adverse reaction reports are voluntary and from a population of unknown size, it is not possible to reliably estimate their frequency.
Table 3
| MedDRA System Organ Class |
Adverse Reaction |
Frequency |
| Blood and lymphatic system disorders |
Development of factor VIII inhibitors |
Uncommon (PLR) ** Very common (PLN) ** |
| Immune system disorders |
Hypersensitivity |
Not known |
| Allergic reactions (hypersensitivity)* |
Not known |
|
| Anaphylactic reaction |
Not known |
|
| Anaphylactic shock |
Not known |
|
| Psychiatric disorders |
Restlessness |
Not known |
| Nervous system disorders |
Headache |
Not known |
| Lethargy |
Not known |
|
| Paraesthesia |
Not known |
|
| Cardiac disorders |
Tachycardia |
Not known |
| Vascular disorders |
Flushing |
Not known |
| Hypotension |
Not known |
|
| Respiratory, thoracic and mediastinal disorders |
Wheezing* |
Not known |
| Gastrointestinal disorders |
Nausea |
Not known |
| Vomiting |
Not known |
|
| Skin and subcutaneous tissue disorders |
Angioedema |
Not known |
| Generalized urticaria (urticaria)* |
Not known |
|
| Rash (urticaria)* |
Not known |
|
| General disorders and administration site conditions |
Burning sensation at infusion site (infusion site pain)* |
Not known |
| Acute pain at infusion site (infusion site pain)* |
Not known |
|
| Chills |
Not known |
|
| Chest tightness (chest discomfort)* |
Not known |
|
| Pyrexia |
Not known |
* MedDRA lower level terms that best describe these adverse reactions; the preferred MedDRA term is provided in parentheses.
** Frequency data are based on results from all Factor VIII product studies involving patients with severe haemophilia A. PEP – previously treated patients; PUP – previously untreated patients.
Children
Specific data in children are lacking.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorization of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Medical and pharmaceutical professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at the following link: https://aisf.dec.gov.ua.
Shelf life
3 years.
Solvent (water for injections) – 5 years.
The medicinal product should be used immediately after reconstitution.
Storage conditions
Store in a refrigerator (at 2 to 8 ℃). Do not freeze. Store in the original packaging to protect from light, in a place inaccessible to children.
Prior to use and within its shelf life, the vial containing the powder may be stored at room temperature not exceeding 25 ℃ for up to 6 consecutive months. After this period, the vial with powder must be destroyed.
After storage of the powder vial at room temperature, further storage in the refrigerator is prohibited.
The date when storage of the medicinal product at room temperature was initiated should be indicated on the original packaging of the medicinal product in the designated space.
Incompatibilities
Due to the lack of compatibility studies, this medicinal product must not be mixed with other medicinal products.
Only the provided injection/infusion set should be used, as treatment may be ineffective due to adsorption of human blood coagulation factor VIII onto the internal surfaces of another administration device.
Packaging
Each pack contains one vial with 500 IU or 1000 IU, one vial with 10 ml of solvent (water for injections), and a reconstitution and administration set, all contained in a cardboard box.
Prescription category: Prescription only.
Manufacturer:
KEDRION S.P.A.
Manufacturer's address and location of operations:
VIA PROVINCIALE (loc. BOLOGNANA) - 55027 GALLICANO (LU), Italy