Somavert
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT SOMAVERT (SOMAVERT)
Composition:
Active substance: pegvisomant;
1 vial contains 10 mg, 15 mg, 20 mg, or 30 mg of pegvisomant;
Excipients: glycine, mannitol (E 421), sodium phosphate anhydrous, sodium dihydrogen phosphate monohydrate.
1 pre-filled syringe with solvent contains water for injections.
Pharmaceutical form. Lyophilisate and solvent for solution for injection.
Main physico-chemical properties: lyophilized mass of white to almost white color.
Pharmacotherapeutic group. Other anterior pituitary hormones and analogues. Pegvisomant. ATC code H01A X01.
Pharmacological Properties
Description
Somavert contains pegvisomant, an analogue of human growth hormone that has been structurally modified to act as a growth hormone receptor antagonist.
Pegvisomant is a protein produced using recombinant DNA technology, consisting of a 191-amino acid residue to which several polyethylene glycol (PEG) polymers are covalently attached (predominantly 4–6 PEG molecules per protein). The molecular mass of the pegvisomant protein is 21,998 daltons. The molecular mass of the PEG portion of pegvisomant is approximately 5,000 daltons. Thus, the predominant molecular mass of pegvisomant is approximately 42,000; 47,000; and 52,000 daltons. Pegvisomant is produced by a specific strain of Escherichia coli bacteria genetically modified by the addition of a plasmid carrying the gene for the growth hormone receptor antagonist. Biological activity is determined by a cell proliferation bioassay. Binding of Somavert to the growth hormone receptor disrupts proper binding with a second growth hormone receptor, thereby inhibiting dimerization of the functional receptor and subsequent intracellular signaling pathway.
Pharmacodynamics
Pegvisomant selectively binds to the growth hormone receptor on the cell surface and has no cross-reactivity with 19 other cytokine receptors studied, including prolactin. Pegvisomant leads to a reduction in serum concentrations of insulin-like growth factor I (IGF-1), free IGF-1, acid-labile subunit (ALS), and insulin-like growth factor binding protein type 3 (IGFBP-3) (see below "Clinical Studies", Fig. 1).
Mechanism of Action
Pegvisomant selectively binds to growth hormone receptors on the cell surface, where it blocks the binding of endogenous growth hormone and thereby prevents growth hormone signal transduction.
Inhibition of growth hormone activity results in decreased serum concentrations of IGF-1, as well as other serum proteins sensitive to growth hormone, such as free IGF-1, acid-labile subunit of IGF-1, and IGFBP-3.
Clinical Studies
A total of 112 patients (63 men and 49 women) with acromegaly participated in a 12-week, randomized, double-blind, multicenter study comparing placebo and Somavert. The mean age ± standard deviation (SD) was 48 ± 14 years, and the mean duration of acromegaly was 8 ± 8 years. Of these patients, 93 had previously undergone pituitary surgery, with 57 of them also receiving standard radiation therapy. Six patients received radiation without prior surgery, 9 received only medical therapy, and 4 patients had not previously received any form of therapy. At the start of the study, the mean ± SD time since the last surgery and/or radiation was 6.8 ± 0.93 years (n = 63) and 5.6 ± 0.57 years (n = 93), respectively.
Patients were included in the study if their serum IGF-1 level, measured after an appropriate washout period, was ≥ 1.3 times above the upper limit of normal, adjusted for age. At the screening visit, patients were randomly assigned to one of four treatment groups: placebo (n = 32); Somavert 10 mg subcutaneously daily (n = 26); Somavert 15 mg subcutaneously daily (n = 26); or Somavert 20 mg subcutaneously daily (n = 28). The primary efficacy endpoint was the percentage change in IGF-1 concentration from baseline at screening to week 12. Statistically significant (p < 0.01) reductions in serum IGF-1 levels were observed in all three Somavert treatment groups compared to the placebo group (Table 1).
Table 1. Mean percentage change in IGF-1 concentration from baseline at screening to week 12 in the "treated patients" population
| Parameters |
Placebo n = 31 |
Somavert |
||
| 10 mg/day n = 26 |
15 mg/day n = 26 |
20 mg/day n = 28 |
||
| Mean IGF-1 concentration at baseline visit (ng/mL) (BV) |
670 (288) |
627 (251) |
649 (293) |
732 (205) |
| Mean percent change in IGF-1 concentration from baseline visit (BV) |
|
|
|
|
| Somavert minus placebo (95% CI for treatment difference) |
(–35; –11) |
(–56; –33) |
(–68; –49) |
|
* P < 0.01; n – number of patients; SD – standard deviation.
Also, in the groups receiving Somavert, a decrease in serum levels of free IGF-1, IGFBP-3, and ALS was observed compared to the placebo group at all scheduled study visits (Fig. 1).
| SOMAVERT 15 mg/day (n = 24–26) SOMAVERT 20 mg/day (n = 27–28) |
| 800 600 400 200 |
| Placebo (n = 31) SOMAVERT 10 mg/day (n = 25–26) |
| 0 2 4 8 12 Week |
| 0 2 4 8 12 Week |
Fig. 1. Effect of Somavert on serum markers (mean ± standard error of the mean).
After 12 weeks of therapy, normalized IGF-1 concentrations were observed in the percentage of patients indicated below (Fig. 2).
| SOMAVERT 20 mg/day |
| SOMAVERT 15 mg/day |
| SOMAVERT 10 mg/day |
| Placebo |
Fig. 2. Percentage of patients in whom IGF-1 concentrations normalized by week 12 of treatment.
Table 2 shows the effect of therapy with the drug Somavert on ring size (standard jewelry sizes converted into a numerical scale from 1 to 63 points) as well as on signs and symptoms of acromegaly. Each individual sign or symptom of acromegaly (soft tissue swelling, arthralgia, headache, sweating, and increased fatigue) was assessed using a nine-point ordinal scale (0 – absent, 8 – severe and disabling), and the total score for evaluating signs and symptoms of acromegaly was calculated as the sum of scores on individual scales. Mean baseline scores were as follows: ring size – 47.1; total score for signs and symptoms – 15.2; soft tissue swelling – 2.5; arthralgia – 3.2; headache – 2.4; sweating – 3.3; and increased fatigue – 3.7.
Table 2. Mean change in ring size and signs and symptoms of acromegaly at week 12 compared to baseline visit (BV).
| Parameters |
Placebo n = 30 |
Somavert |
||
| 10 mg/day n = 26 |
15 mg/day n = 24–25 |
20 mg/day n = 26–27 |
||
| Ring size |
|
|
|
|
| Total symptom score for acromegaly signs and symptoms |
1.3 (6.0) |
|
|
|
| Soft tissue swelling |
0.3 (2.3) |
|
|
|
| Arthralgia |
0.1 (1.8) |
|
|
|
| Headache |
0.1 (1.7) |
|
|
|
| Sweating |
0.1 (1.7) |
|
|
|
| Increased fatigue |
0.7 (1.5) |
|
|
|
Serum growth hormone concentrations, measured using quantitative assay methods developed for the study and employing antibodies without cross-reactivity to pegvisomant, increased within two weeks after initiation of therapy with Somavert. The greatest increase in growth hormone concentration was observed in patients receiving Somavert at a dose of 20 mg/day. This effect is most likely due to reduced inhibition of growth hormone secretion resulting from decreased IGF-1 levels. As shown in Fig. 3, when patients with acromegaly received a loading dose of Somavert followed by fixed daily doses, the increase in growth hormone concentration was inversely proportional to the decrease in IGF-1 levels and typically stabilized by week 2. Serum growth hormone concentrations remained stable in patients receiving Somavert for a mean duration of 43 weeks (range 0–82 weeks).
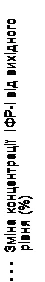
| Placebo |
| Week Week Week Week Week 0 2 4 8 12 |
| 20 mg/day |
| 15 mg/day |
| 10 mg/day |

Fig. 3. Percentage change in serum growth hormone and IGF-1 concentrations.
In the open-label extension phase of the clinical study, 109 patients participated (including 6 newly enrolled patients), with a mean duration of treatment of 42.6 weeks (range: 1 day – 82 weeks). Adverse reactions occurred in 93 (85.3%) patients; serious adverse reactions (SARs) were observed in 16 (14.7%) patients, and treatment was discontinued due to adverse reactions in 4 (3.7%) patients (headache, increased liver function test levels, pancreatic cancer, and weight gain). Overall, 100 (92.6%) of 108 participants with available IGF-1 measurements had normal IGF-1 concentrations at any visit during the study.
In various studies, including the ACROSTUDY trial, pegvisomant normalized IGF-1 levels in a large proportion of patients (> 70%) and significantly reduced fasting plasma glucose (FPG) and fasting plasma insulin (FPI) levels.
Pegvisomant also improves insulin sensitivity, which is likely related to blockade of GH receptors in tissues, primarily in the liver, as well as adipose tissue, kidneys, and skeletal muscle, thereby eliminating the harmful effects of GH on insulin signaling pathways, lipolysis, and gluconeogenesis. However, the mechanism of this effect is not well established. Diabetic patients with acromegaly may require dose reductions of insulin or antihyperglycemic agents (see sections "Special warnings and precautions for use", "Interaction with other medicinal products and other forms of interaction").
Pharmacokinetics.
Absorption. Following subcutaneous administration, peak serum concentrations of pegvisomant are typically achieved no earlier than 33–77 hours after dosing. The mean extent of absorption of a 20 mg subcutaneous dose is 57% compared to that after a 10 mg intravenous dose.
Distribution. The mean apparent volume of distribution of pegvisomant is 7 L (coefficient of variation 12%), indicating minimal tissue distribution. After single subcutaneous administration, exposure (Cmax, AUC) of pegvisomant increases disproportionately with increasing dose. The mean ± standard error of the mean (SEM) serum concentration of pegvisomant after 12 weeks of daily doses of 10, 15, and 20 mg was 6600 ± 1330, 16,000 ± 2200, and 27,000 ± 3100 ng/mL, respectively.
The relative bioavailability of a single 30 mg dose of pegvisomant was compared to two 15 mg doses in a single-dose study. The AUCinf and Cmax values of pegvisomant following administration of a single 30 mg injection were approximately 6% and 4% higher, respectively, than those following two 15 mg injections.
Metabolism and elimination. The pegvisomant molecule contains covalently bound polyethylene glycol polymers designed to reduce clearance rate. The clearance of pegvisomant after multiple dosing is lower than that observed after single dosing. The mean total systemic clearance of pegvisomant in the body after multiple dosing is estimated to range from 36 to 28 mL/h for subcutaneous doses ranging from 10 to 20 mg per day, respectively. Pegvisomant clearance has been found to increase with body weight. Pegvisomant is eliminated from serum with a mean half-life ranging from 60 to 138 hours after single or multiple administrations. Less than 1% of the administered dose is recovered in urine within 96 hours. The elimination pathways of pegvisomant in humans have not been studied.
Drug interaction studies
In clinical trials, patients receiving opioids often required higher serum concentrations of pegvisomant to achieve adequate IGF-1 suppression compared to patients not receiving opioids. The mechanism of this interaction is unknown (see section "Interaction with other medicinal products and other forms of interaction").
Special patient groups
Pharmacokinetic studies in patients with impaired renal function, hepatic impairment, elderly patients, or pediatric patients have not been conducted. The effect of patient race on pegvisomant pharmacokinetics has not been studied. Population pharmacokinetic analysis revealed no differences in pegvisomant pharmacokinetics based on patient sex.
Immunogenicity
The observed frequency of antibodies to the drug is highly dependent on the sensitivity and specificity of the analytical method. Differences in analytical methods preclude meaningful comparison of antibody frequencies across the studies described below with those from other studies, including studies of Somavert or other growth hormone analogs.
In pre-approval clinical trials, approximately 17% of patients receiving Somavert developed low titers of neutralizing antibodies to growth hormone. Although the presence of these antibodies appears not to affect the efficacy of Somavert, the long-term clinical significance of their development is unknown. Currently, there are no commercially available diagnostic kits for detecting antibodies to pegvisomant in patients receiving Somavert.
The data presented above reflect the percentage of patients with positive test results for antibodies to Somavert. Detection of antibody formation is highly dependent on the sensitivity and specificity of the assay method. Furthermore, the observed frequency of positive antibody test results may be influenced by several factors, including sample handling, timing of sample collection, concomitant medications, and underlying disease. Therefore, comparisons of the incidence of antibody formation to Somavert with that of other drugs may yield misleading results.
Clinical characteristics.
Indications.
Somavert is indicated for the treatment of acromegaly in patients who have had an inadequate response to surgery or radiotherapy, or for whom these therapies are not suitable. The goal of treatment is to normalize serum insulin-like growth factor-1 (IGF-1) levels.
Contraindications.
Somavert must not be used in patients who are allergic to the active substance or to any of the other ingredients of the medicinal product.
Interaction with other medicinal products and other forms of interaction.
Insulin and/or oral hypoglycemic agents
After initiation of Somavert therapy, patients with acromegaly and diabetes mellitus who are receiving insulin and/or oral hypoglycemic agents may require a reduction in the dose of insulin and/or oral hypoglycemic agents (see section "Dosage and administration").
Opioids
In clinical studies, patients receiving opioids often required higher doses of Somavert to normalize IGF-1 concentrations than patients not receiving opioids. The mechanism of this interaction is unknown.
Special precautions for use
Hypoglycemia associated with decreased growth hormone levels in diabetic patients
Growth hormone counteracts the effects of insulin on carbohydrate metabolism by reducing insulin sensitivity. Thus, in some patients receiving Somavert, glucose tolerance may improve. Close monitoring of patients with diabetes mellitus is required, and if necessary, doses of antidiabetic agents should be reduced to avoid hypoglycemia.
Hepatotoxicity
Prior to initiating therapy with Somavert, baseline assessment of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), total bilirubin, and alkaline phosphatase levels should be performed. Table 3 provides recommendations for initiating Somavert therapy based on baseline liver function test (LFT) results.
Asymptomatic, transient elevations in transaminase levels up to 15 times the upper limit of normal (ULN) were observed in < 2% of patients in two open-label studies (involving a total of 147 patients). These elevated transaminase levels were not accompanied by increases in bilirubin. Elevated transaminase levels returned to normal over time, usually after temporary discontinuation of therapy. Post-marketing reports have identified increases in serum hepatic transaminases to more than 20 times the ULN, associated with elevations in total bilirubin exceeding twice the UL, in some cases. In many of these cases, discontinuation of Somavert therapy resulted in reduction or normalization of abnormal liver parameters.
During therapy, Somavert should be used according to the information regarding abnormalities in liver biochemical parameters as outlined in Table 4.
Table 3. Recommendations for initiating Somavert therapy based on baseline liver biochemical parameters and for periodic monitoring of liver biochemical parameters during Somavert therapy
| Baseline liver function tests |
Recommendations |
| Normal |
|
| Elevated, but ≤ 3 × ULN |
Pegvisomant may be used; however, liver function tests should be monitored monthly for at least one year after initiation of therapy, then twice yearly for the following year. |
| More than 3 times ULN |
|
If during treatment with Somavert a patient's GGT level increases or any other signs or symptoms of hepatic dysfunction appear, the management outlined below is recommended (Table 4).
Table 4. Clinical recommendations based on assessment of liver function biochemical parameters during treatment with Somavert
| ALT level and clinical signs/symptoms |
Recommendations |
| Greater than or equal to 3 × ULN but less than 5 × ULN (without signs/symptoms of hepatitis or other liver injury or without increased serum total bilirubin) |
|
| At least 5 times higher than ULN or transaminase levels at least 3 times higher than ULN accompanied by any increase in serum total bilirubin (with or without signs/symptoms of hepatitis or other liver injury) |
|
| Signs or symptoms indicating hepatitis or other liver injury (e.g., jaundice, bilirubinuria, increased fatigue, nausea, vomiting, right upper quadrant abdominal pain, ascites, oedema of unknown aetiology, tendency to bruising) |
|
Patients should have blood drawn to analyze IGF-1 levels and liver function biochemical markers before and during treatment with Somavert; the dose of the drug may be adjusted based on the results of these tests.
Cross-reactivity with quantitative growth hormone assays
Somavert has significant structural similarity to growth hormone, resulting in cross-reactivity when using quantitative growth hormone assay kits. Since the concentration of Somavert in serum during administration of therapeutically effective doses is typically 100–1000 times higher than the actual concentration of growth hormone in serum of patients with acromegaly, measurements of serum growth hormone levels may yield falsely elevated values.
Growth of growth hormone-secreting tumors
Pituitary tumors producing growth hormone may occasionally grow and cause serious complications (e.g., visual field defects); therefore, it is important that all patients be closely monitored. If signs of tumor growth occur, alternative procedures may be recommended.
Lipohypertrophy
Cases of lipohypertrophy have been observed in patients using Somavert. In a double-blind, 12-week, placebo-controlled study, one case (1.3%) of injection-site lipohypertrophy was reported in a patient receiving 10 mg/day. The condition resolved while continuing therapy. In two open-label studies (involving a total of 147 patients), two patients (both receiving 10 mg/day) developed lipohypertrophy. In one case, the condition resolved during continued treatment, while in the other it led to discontinuation of therapy. To prevent lipohypertrophy, the injection site should be rotated daily (i.e., the drug should be administered at a different site than the previous injection).
Renal impairment
Somavert has not been studied in patients with impaired renal function, and the safety and efficacy of the drug in such patients are unknown.
Use in elderly patients
The number of participants aged 65 years and older in clinical studies of Somavert was not sufficient to determine whether their response differs from that of younger patients. In general, dose selection for elderly patients should be cautious, usually starting at the lower end of the dosing range, due to the higher likelihood of decreased hepatic, renal, or cardiac function and the presence of concomitant diseases or other drug therapies in this age group.
Sodium content
This medicinal product contains less than 1 mmol of sodium (23 mg) per dose. Patients on a low-sodium diet may be informed that this medicinal product can be considered essentially sodium-free.
Use during pregnancy or breastfeeding.
Pregnancy
Overview of risk-related information
There are insufficient postmarketing data on the use of Somavert in pregnant women to determine a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. The clinical course of acromegaly may improve during pregnancy (see section "Overview of clinical data"). In animal reproduction studies, fetotoxicity was observed at a dose 6 times the maximum recommended human dose based on body surface area, following subcutaneous administration of pegvisomant during organogenesis or the pre-implantation period (see section "Data"). The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Overview of clinical data
Maternal and/or embryofetal risk associated with the disease
Published case reports, case series, and a small interventional study in pregnant women with acromegaly have shown that the clinical course of acromegaly may improve or stabilize without treatment during pregnancy, especially if acromegaly was controlled prior to pregnancy. In rare cases, acromegaly may worsen during pregnancy. Because hormone levels may change physiologically during pregnancy and interpretation of growth hormone levels in pregnant women with acromegaly may be unreliable, clinical monitoring is recommended. The use of the drug in pregnant women has not been studied; therefore, patients should inform their physician immediately if they become pregnant.
Data
Animal reproduction studies data
The effects of pegvisomant on early embryonic and embryofetal development were evaluated in two separate studies in pregnant rabbits receiving subcutaneous pegvisomant at doses of 1, 3, and 10 mg/kg/day. There was no evidence of teratogenic effects associated with pegvisomant administration during organogenesis. At the dose of 10 mg/kg/day (6 times the maximum recommended human dose based on body surface area), a reproducible slight increase in post-implantation loss was observed in both studies.
Breastfeeding
Overview of risk-related information
According to limited clinical case information, published reports indicate that pegvisomant levels in human breast milk were below the level of detection. There is no available information on the effect of the drug on the breastfed infant or on milk production. The benefits of breastfeeding for the child's development and health, the mother's clinical need for Somavert, and any potential adverse effects of Somavert on the breastfed child or on the mother's underlying condition should be considered.
A patient should inform her physician if she plans to breastfeed.
Women and men of reproductive potential
The possibility of unexpected pregnancy should be discussed with premenopausal women, as the therapeutic benefits of lowering growth hormone (GH) levels and normalizing insulin-like growth factor 1 (IGF-1) concentrations in women with acromegaly receiving pegvisomant may lead to improved fertility.
Ability to affect reaction speed when driving or operating machinery.
Studies on the effect of the drug on the ability to drive vehicles or operate machinery have not been conducted.
Administration and Dosage
Dosing Information
The recommended loading dose of Somavert is 80 mg administered subcutaneously under medical supervision. Proper instruction on the technique for performing subcutaneous injections must be provided. Starting the day after administration of the loading dose, the patient should receive daily subcutaneous injections of Somavert at a dose of 10 mg.
The dose should then be titrated to normalize serum IGF-1 concentration (serum IGF-1 levels should be measured every 4–6 weeks). Dose selection should not be based on growth hormone concentration or signs and symptoms of acromegaly. It is unknown whether patients who still have symptoms after achieving normal IGF-1 levels would benefit from increasing the dose of Somavert.
-
If IGF-1 concentration is elevated, the dose of the drug should be increased by 5 mg every 4–6 weeks.
-
If IGF-1 concentration is below the normal range, the dose of the drug should be decreased by 5 mg every 4–6 weeks.
-
IGF-1 levels should also be monitored when switching a patient from a daily regimen of Somavert administered as multiple injections to a daily regimen with the required dose given as a single injection (see section "Pharmacological Properties").
The recommended dosage range is 10 to 30 mg subcutaneously once daily. The maximum daily dose is 30 mg subcutaneously once daily.
Assessment of Hepatic Function Parameters Prior to Initiation of Somavert Therapy
Prior to initiating treatment with Somavert, baseline assessment of liver function tests should be performed (serum alanine aminotransferase [ALT], aspartate aminotransferase [AST], total serum bilirubin [TSB], and alkaline phosphatase). Recommendations for initiating Somavert therapy based on baseline liver function parameters and monitoring recommendations during Somavert therapy are provided in Table 3 (see section "Special Warnings and Precautions for Use").
Procedure for Administration of the Loading Dose Injection
The instructions below regarding reconstitution and preparation for administration of the 80 mg loading dose are intended for healthcare professionals. The healthcare professional must reconstitute the lyophilized powder in 4 vials of Somavert, each containing 20 mg of pegvisomant, using the diluent supplied in the package (4 vials of lyophilized powder and 4 syringes containing 2.25 mL diluent [sterile water for injection] are required to prepare the 80 mg loading dose). The healthcare professional must also administer 4 injections of the reconstituted Somavert solution at sites selected from the patient's shoulder, upper thigh, abdomen, or buttock (each injection at a different site).
- Prior to administration of the loading dose, remove the first package (1 vial of Somavert lyophilized powder containing 20 mg pegvisomant and 1 syringe containing 2.25 mL diluent) from the refrigerator approximately 10 minutes before the scheduled injection time.
- Reconstitute the 20 mg pegvisomant lyophilized powder in the first vial with the provided diluent. When using the 2.25 mL syringe of diluent, slowly inject the contents along the wall of the vial containing the Somavert lyophilized powder. Do not direct the stream of diluent directly onto the powder.
- Do not invert or shake the vial, as this may cause denaturation of the pegvisomant protein. Gently mix the solution by swirling to ensure complete dissolution of the lyophilized powder. If foaming is observed in the reconstituted Somavert solution, the solution may be compromised and therefore unsuitable for administration.
- Visually inspect the reconstituted Somavert solution for particulate matter and discoloration prior to administration. The reconstituted solution should be clear and free of visible particles. Do not use the solution if it is cloudy. After reconstitution, the solution contains 20 mg of pegvisomant per 1 mL.
- Draw up 1 mL of the reconstituted Somavert solution. The solution must be used within 6 hours after reconstitution.
- Administer the first subcutaneous injection of the reconstituted Somavert solution (20 mg/mL) into the patient’s shoulder, upper thigh, abdomen, or buttock at a 90-degree angle.
- Repeat steps (a) through (d) to reconstitute the second, third, and fourth 20 mg doses of Somavert.
- Then administer the second (followed by the third and fourth) subcutaneous injection of the reconstituted Somavert solution (20 mg/mL) into the patient’s shoulder, upper thigh, abdomen, or buttock (select a different site than previous injections) at a 90-degree angle.
- Repeat steps (a) through (d) to reconstitute the second, third, and fourth 20 mg doses of Somavert.
- Administer the first subcutaneous injection of the reconstituted Somavert solution (20 mg/mL) into the patient’s shoulder, upper thigh, abdomen, or buttock at a 90-degree angle.
- Draw up 1 mL of the reconstituted Somavert solution. The solution must be used within 6 hours after reconstitution.
- Visually inspect the reconstituted Somavert solution for particulate matter and discoloration prior to administration. The reconstituted solution should be clear and free of visible particles. Do not use the solution if it is cloudy. After reconstitution, the solution contains 20 mg of pegvisomant per 1 mL.
- Do not invert or shake the vial, as this may cause denaturation of the pegvisomant protein. Gently mix the solution by swirling to ensure complete dissolution of the lyophilized powder. If foaming is observed in the reconstituted Somavert solution, the solution may be compromised and therefore unsuitable for administration.
- Reconstitute the 20 mg pegvisomant lyophilized powder in the first vial with the provided diluent. When using the 2.25 mL syringe of diluent, slowly inject the contents along the wall of the vial containing the Somavert lyophilized powder. Do not direct the stream of diluent directly onto the powder.
Procedure for Administration of the Maintenance Dose Injection
Instructions for the patient or caregiver on reconstitution and administration of daily doses (ranging from 10 to 30 mg) are provided below in the "Step-by-Step Instructions for Administration of Somavert."
- Prior to administration of the dose, remove one vial of Somavert lyophilized powder containing 10 mg, 15 mg, 20 mg, or 30 mg pegvisomant and one 2.25 mL syringe containing diluent from the refrigerator approximately 10 minutes before the scheduled injection time.
- Reconstitute the Somavert lyophilized powder using the diluent. When using the 2.25 mL syringe of diluent, slowly inject the contents along the wall of the vial containing the Somavert lyophilized powder. Do not direct the stream of diluent directly onto the powder.
- Do not invert or shake the vial, as this may cause denaturation of the pegvisomant protein. Gently mix the solution by swirling to ensure complete dissolution of the lyophilized powder. If foaming is observed in the reconstituted Somavert solution, the solution may be compromised and therefore unsuitable for administration.
- Prior to administration, visually inspect the reconstituted Somavert solution for particulate matter and discoloration. The reconstituted solution should be clear. Do not use the solution if it is cloudy. After reconstitution, the solution contains 10 mg, 15 mg, 20 mg, or 30 mg of pegvisomant per 1 mL.
- Draw up 1 mL of the reconstituted Somavert solution. The solution must be used within 6 hours after reconstitution.
Administer the subcutaneous injection of the reconstituted Somavert solution into the shoulder, upper thigh, abdomen, or buttock at a 90-degree angle.
Step-by-Step Instructions for Administration of Somavert
Step 1. Supplies Needed
One package of Somavert contains a vial of Somavert powder, a pre-filled syringe, and a safety needle.
You will also need: a cotton swab, an alcohol wipe, and a sharps disposal container.
| finger flange on the syringe |
| flacon stopper (with removed flacon cap) |
| needle cap |
| Safety needle |
| Syringe |
| protective needle cover |
| syringe plunger rod |
| syringe tip |
| syringe cylinder |
| syringe cap |
| expiry date |
| flacon cap |
| opening in the stopper |
| Bottle |
Step 2. Preparation
Before starting:
- Mix Somavert and diluent only when you are ready to administer the dose.
- Remove one package of Somavert from the refrigerator and allow the medication to warm to room temperature in a safe place for at least 10 minutes before use.
- Do not warm the Somavert package using a heat source, such as hot water or a microwave oven. The medication should warm on its own.
- Wash your hands with soap and water and dry them completely.
- Open the package containing the syringe and the safety needle, arranging all necessary items within easy reach for preparing the injection.
- Do not use syringes or vials if:
- they are damaged or defective;
- the expiration date has passed;
- they have been frozen, even if they are now thawed (this applies only to syringes).
Step 3. Choosing the injection site
| Selection of injection site |
| Abdomen Stay at least 5 cm away from the navel |
| Thighs |
| Shoulder or lower back Back of the upper arm (for healthcare provider or caregiver only) |
- For each injection, select a different site within the injection areas.
- Avoid areas near bones or areas with bruises, redness, pain, or lumps, as well as areas with scars or skin conditions.
- Wipe the injection site with an alcohol swab.
- Allow the injection site to dry.
Step 4. Removing the cap from the vial
| Removing the cap from the vial |
- Remove the cap from the vial.
- Discard the cap. You will not need it anymore.
Caution! Do NOT touch the vial stopper with anything.
Step 5. Removing the syringe cap
| Removing the cap from the syringe |
| Clicking |
- Remove the cap from the syringe, leaving the syringe tip on. You may need to use more force than expected to remove the cap.
- Discard the syringe cap. It will not be needed anymore.
- Hold the syringe upright to prevent leakage of liquid.
Caution! Do not touch the syringe tip to any surfaces once the cap has been removed.
Step 6. Attaching the safety needle
| Attaching |
- Press and twist the safety needle firmly onto the syringe until it stops.
Step 7. Removing the needle cap
| Removing the needle cap |
- Remove the protective needle cover from the path of the needle cap.
- Carefully remove the cap from the needle.
- Discard the needle cap. You will not need it anymore.
Caution! Do not let the needle touch anything.
Step 8. Inserting the needle
| Needle insertion |
- Pierce the center of the vial stopper with the needle, as shown in the figure.
- Hold the syringe steady while the needle remains in the vial stopper to prevent bending the needle.
Step 9. Adding the liquid
| Addition of liquid |
- Tilt the vial and syringe at an angle, as shown in the illustration.
- Slowly press the syringe plunger until all the liquid has transferred into the vial.
- Caution! Do not direct the liquid stream directly onto the powder. This may cause foaming, rendering the preparation unsuitable for use.
- Do not remove the needle yet.
Step 10. Mix the solution by rotating in circular motions
| Mixing the solution with circular movements |
- Hold the syringe and vial in one hand, as shown in the illustration.
- Carefully and slowly mix the solution by moving the vial in a circular motion on a flat surface.
- Continue mixing the liquid with circular movements until all the powder is completely dissolved.
Note: This may take up to 5 minutes. Do not shake.
Step 11. Check the solution
| Inspection of the solution |
- While holding the needle in the vial, carefully examine the solution. It should be clear and free of foreign particles.
- Do not use the medication if:
- the solution is cloudy or discolored;
- the solution is colored in any way;
- there are any particles or foam present in the vial.
Step 12. Moving the needle
| Needle movement |
- Turn the vial upside down so that you can see the opening in the stopper, as shown in the figure.
- Position the needle downward so that the tip of the needle is in the lowest point of the liquid. This will allow you to withdraw as much liquid from the vial as possible.
- Make sure the syringe plunger is not moving. If the syringe plunger has moved, push it back into the syringe. This ensures removal of all air from the syringe before you begin drawing up the dose.
Step 13. Drawing up the dose
| Dose titration |
- Slowly pull back the syringe plunger to withdraw as much solution as possible from the vial.
Note: If you see air bubbles in the syringe, gently tap the syringe barrel to allow the bubbles to rise to the top, then carefully expel the air bubbles back into the vial.
- Remove the needle from the vial.
Step 14. Inserting the needle
| Needle insertion |
- Gently grasp the skin fold at the injection site.
- Insert the needle fully into the skin fold.
Step 15. Administering the medication injection
| Administration of the drug injection |
- Slowly push the syringe plunger down until the syringe cylinder is empty.
Note. Make sure you have inserted the needle fully.
- Release the skin fold and withdraw the needle at a right angle.
Step 16. Needle safety
| Click! |
| Needle safety |
- Lower the needle guard over the needle.
Carefully press down on the guard using a hard surface to return it to its original position.
- Note: You will hear a click when the protective guard locks into place.
Step 17. Disposal
- Place used syringes into a sharps disposal container.
- Do not dispose of (do not throw away) syringes in household waste.
Step 18. After the injection
- If needed, take a clean cotton pad and gently press it against the injection site.
- Do not rub the injection site.
Children
The safety and efficacy of Somavert in children have not been established.
Overdose
During post-marketing clinical experience, one case of acute overdose with Somavert has been reported: a patient self-administered injections at a dose of 80 mg/day (2.7 times higher than the maximum recommended maintenance dose) for seven days. The patient experienced mild fatigue without any other complaints or clinically significant abnormalities in laboratory test results.
In case of overdose, Somavert administration should be discontinued and not resumed until IGF-1 levels return to normal.
Adverse Reactions
Clinically significant adverse reactions mentioned in the section "Special Warnings and Precautions for Use" include:
- Hypoglycemia associated with decreased growth hormone levels in patients with diabetes mellitus;
- Hepatotoxicity;
- Cross-reactivity in growth hormone immunoassays;
- Lipohypertrophy;
- Systemic hypersensitivity reactions.
During pre-approval clinical trials, serum ALT and AST concentrations increased more than 10 times above the upper limit of normal (ULN) in two patients (0.8%) receiving Somavert. One patient underwent a rechallenge with Somavert, after which there was a recurrence of elevated transaminases, indicating a probable causal relationship between drug administration and increased liver enzymes. Liver biopsy findings in the second patient were consistent with chronic hepatitis of unknown etiology. In both patients, elevated transaminase levels returned to normal after discontinuation of the drug.
Elevations in ALT and AST were not accompanied by increases in total bilirubin or alkaline phosphatase, except in two patients who had minimal associated increases in alkaline phosphatase (i.e., less than 3 × ULN). Transaminase elevations are likely not dose-dependent and typically occur within 4–12 weeks after initiation of therapy. No biochemical, phenotypic, or genetic predictive factors associated with this phenomenon have been identified.
Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, the frequency of adverse reactions observed in the clinical trials of one drug cannot be directly compared to the frequency in clinical trials of another drug, and may not reflect the frequency observed in practice.
In a 12-week randomized, placebo-controlled, double-blind, fixed-dose study of Somavert in patients with acromegaly, 32 patients received placebo and 80 patients received Somavert once daily (see section "Pharmacological Properties" ("Clinical Studies")). Overall, 108 patients (30 in the placebo group, 78 in the Somavert group) completed 12 weeks of therapy.
Overall, eight patients with acromegaly (5.3%) were withdrawn from pre-approval clinical trials due to adverse reactions, including two patients with marked elevations in transaminase levels, one patient with injection-site lipohypertrophy, and one patient with significant weight gain. Most adverse reactions appeared to be non-dose-dependent. Table 5 lists adverse reactions reported in at least two patients receiving Somavert and occurring at a higher frequency than in the placebo group during the 12-week placebo-controlled study.
Table 5. Adverse Reactions Observed in the 12-Week Placebo-Controlled Study in Patients with Acromegaly*
Adverse Reactions |
Placebo n = 32 |
Somavert |
||
| 10 mg/day n = 26 |
15 mg/day n = 26 |
20 mg/day N = 28 |
||
| Infection† |
2 (6 %) |
6 (23 %) |
0 |
0 |
| Pain |
2 (6 %) |
2 (8 %) |
1 (4 %) |
4 (14 %) |
| Nausea |
1 (3 %) |
0 |
2 (8 %) |
4 (14 %) |
| Diarrhea |
1 (3 %) |
1 (4 %) |
0 |
4 (14 %) |
| Abnormal liver function biochemical test |
1 (3 %) |
3 (12 %) |
1 (4 %) |
1 (4 %) |
| Influenza-like syndrome |
0 |
1 (4 %) |
3 (12 %) |
2 (7 %) |
| Injection site reaction |
0 |
2 (8 %) |
1 (4 %) |
3 (11 %) |
| Dizziness |
2 (6 %) |
2 (8 %) |
1 (4 %) |
1 (4 %) |
| Accidental injury |
1 (3 %) |
2 (8 %) |
1 (4 %) |
0 |
| Back pain |
1 (3 %) |
2 (8 %) |
0 |
1 (4 %) |
| Sinusitis |
1 (3 %) |
2 (8 %) |
0 |
1 (4 %) |
| Chest pain |
0 |
1 (4 %) |
2 (8 %) |
0 |
| Peripheral edema |
0 |
2 (8 %) |
0 |
1 (4 %) |
| Arterial hypertension |
0 |
0 |
2 (8 %) |
0 |
| Paresthesia |
2 (6 %) |
0 |
0 |
2 (7 %) |
* The table includes only reactions that were reported in at least 2 patients and occurred more frequently in patients receiving Somavert than in placebo group patients.
† In the group receiving Somavert 10 mg, 6 reactions coded as "infection" were reported: cold symptoms (3), upper respiratory tract infection (1), blisters (1), and ear infection (1). In the placebo group, 2 such reactions were recorded: cold symptoms (1) and chest organ infection (1).
Post-marketing experience
Adverse reactions reported from post-marketing spontaneous reports
During the post-marketing period of Somavert use, the following adverse reactions have been identified. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Systemic hypersensitivity reactions, including anaphylactic reactions, laryngospasm, angioedema, and generalized skin reactions (rash, erythema, pruritus, urticaria), have been reported during the post-marketing period. Some patients required hospitalization. Upon re-initiation of therapy, symptoms did not recur in any patient (see section "Special warnings and precautions for use" ("Systemic hypersensitivity reactions")).
Adverse reactions from observational study
ACROSTUDY is an international observational registry collecting long-term safety data in 2221 patients with acromegaly treated with Somavert, with a median treatment duration of 8.5 years. Patients were allowed to receive other acromegaly treatments during the study period. The dose and regimen were determined at the discretion of each prescribing physician. Although safety monitoring according to the recommended schedule was required, not all assessments were performed at all time points for every patient. Therefore, comparison of adverse reaction frequencies with those observed in the original clinical trial is not feasible.
Among 1327 patients with normal baseline AST and ALT levels, elevations >3–5 times above ULN were observed in 20 (1.5%) patients, and elevations >5 times above ULN occurred in 22 (1.7%) patients. Lipohypertrophy was reported in 35 (1.6%) patients.
Among 1795 study participants with available baseline MRI and at least one follow-up MRI, MRI results showed an increase in 128 (7.1%), a decrease in 310 (17.3%), both increase and decrease in 81 (4.5%), and no change in 1276 (71.1%).
Patients (and/or caregivers) should be informed about the potential development of the following adverse reactions:
- The most commonly reported adverse reactions were injection site reactions, increased liver function biochemical parameters, pain, nausea, and diarrhea.
- If patients develop elevated liver function biochemical parameters, they may require more frequent blood testing for liver function parameters and/or discontinuation of Somavert. Inform patients to immediately stop therapy and contact their physician if jaundice occurs.
- In patients with acromegaly, growth hormone-secreting tumors may enlarge and should be carefully monitored by magnetic resonance imaging (MRI).
- Subcutaneous injection site nodules may develop, potentially leading to lipoatrophy; rotating injection sites may help prevent or minimize this occurrence.
- In patients with diabetes, careful monitoring may be required, and doses of insulin and/or oral hypoglycemic agents may need to be reduced during treatment with Somavert.
- Patients receiving opioids may require higher doses of Somavert to achieve adequate IGF-1 inhibition.
Reporting suspected adverse reactions
Reporting suspected adverse reactions after medicine authorization is important. It allows ongoing monitoring of the benefit-risk balance of the medicine. Healthcare professionals, patients, and their legal representatives are encouraged to report all suspected adverse reactions and lack of efficacy through the automated pharmacovigilance information system at: https://aisf.dec.gov.ua.
Shelf life.
3 years.
Storage conditions.
Store vials of lyophilized powder in the original packaging protected from light at 2–8°C in a refrigerator. Do not freeze.
The medicine in vials in original packaging may be stored at temperatures not exceeding 25°C for a single period of up to 30 days. The cardboard box should be marked with the date by which the product must be used (within 30 days from removal from the refrigerator). Vials must be protected from light and must not be returned to the refrigerator. The product should be disposed of if not used within 30 days of storage at room temperature or after the expiry date stated on the box, whichever comes first.
The solvent in pre-filled syringes should be stored at temperatures not exceeding 30°C or at 2–8°C in a refrigerator. Do not freeze.
The reconstituted solution should be used immediately or within 6 hours after preparation.
Keep out of reach of children.
Packaging.
10 vials of lyophilized powder in an intermediate cardboard box; 3 intermediate cardboard boxes, 30 pre-filled syringes with 1 ml solvent, 30 safety needles in a cardboard box.
Prescription status.
Prescription only.
Manufacturer.
Pfizer Manufacturing Belgium NV.
Manufacturer's location and address of operations.
Rijksweg 12, Puurs-Sint-Amands, 2870, Belgium.