Hemax
Ukraine
Table of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT HEMAX (HEMAX)
Composition:
Active substance: erythropoietin (epoetin alfa);
One vial contains erythropoietin (epoetin alfa) 1,000 IU, 2,000 IU, 3,000 IU, 4,000 IU, 10,000 IU, 20,000 IU, or 40,000 IU;
Excipients: mannitol (E 421), sodium chloride, sodium dihydrogen phosphate dihydrate, sodium phosphate dodecahydrate, human albumin.
Pharmaceutical form. Lyophilisate for solution for injection.
Main physicochemical properties: white, homogeneous, dense powder.
Pharmacotherapeutic group. Agents affecting the blood and hematopoietic system. Antianemic agents. Other antianemic agents. Erythropoietin.
ATC code B03XA01.
Pharmacological properties.
Pharmacodynamics.
Hemax is a medicinal product whose active pharmaceutical ingredient is epoetin alfa (recombinant human erythropoietin, r-HuEPO). Epoetin is a glycoprotein containing 165 amino acids, produced using recombinant technology. It is produced by genetically modified mammalian cells containing the erythropoietin gene. The product is highly purified and contains an amino acid sequence identical to that of natural human erythropoietin.
Erythropoietin induces erythropoiesis by stimulating the division and differentiation of erythroid precursor cells, resulting in an increased number of red blood cells and hematocrit. Erythropoietin also stimulates the release of reticulocytes from the bone marrow into the bloodstream, where they mature into erythrocytes. Normally, serum erythropoietin concentration ranges from 10 to 30 IU/mL and is regulated by tissue oxygenation levels. When tissue oxygen levels decrease, erythropoietin concentration increases 100–1000 times. This phenomenon is also observed in anemias.
Pharmacokinetics.
Hemax (active substance – epoetin alfa) is administered parenterally (subcutaneously or intravenously). Reticulocyte count increases within 7–10 days after administration of the drug. Erythrocyte count, hematocrit, and hemoglobin levels typically rise within 2–6 weeks after administration of epoetin alfa. The magnitude and speed of response depend on the dose of the drug and the availability of serum iron stores. Maximum plasma concentration is achieved within 15 minutes after a single intravenous dose and within 5–24 hours after a single subcutaneous dose. Peak serum erythropoietin levels may persist from 12 to 16 hours after subcutaneous administration, and erythropoietin remains detectable in serum 24 hours after administration.
The elimination half-life of epoetin alfa ranges from 4 to 13 hours following intravenous or subcutaneous administration. The half-life after the first dose is longer than after two or more weeks of treatment. Typically, erythropoietin levels return to baseline within 24 hours. After subcutaneous administration, maximum plasma concentration occurs within 5–24 hours, and subsequent decline in concentration proceeds more slowly.
Studies in healthy adult volunteers have shown that the half-life after intravenous administration is 20% shorter than in patients with renal insufficiency. The half-life after subcutaneous administration of Hemax in healthy adult volunteers is 20.8 ± 6.3 hours. After discontinuation of Hemax treatment, hematocrit begins to decrease within 2 weeks.
Clinical Characteristics.
Indications.
- Treatment of symptomatic anemia associated with chronic kidney disease:
- in adults and children on hemodialysis and in adults on peritoneal dialysis;
- in adult patients with kidney insufficiency not yet undergoing hemodialysis, for treatment of severe renal anemia accompanied by clinical symptoms.
- Treatment of anemia and reduction of the need for blood transfusions in adult patients receiving chemotherapy due to solid tumors, malignant lymphoma, or multiple myeloma, who are at increased risk of transfusion as assessed by overall patient condition (including cardiovascular status, pre-existing anemia prior to initiation of chemotherapy).
- Hemax is indicated within a pre-deposit program to facilitate collection of autologous blood in patients with mild anemia (hemoglobin level 10–13 g/dL (6.2–8.1 mmol/L), absence of iron deficiency). Treatment should be prescribed only when blood-sparing procedures are not feasible or insufficient (in cases where major planned surgery is expected to result in significant blood loss – 4 or more units of blood for women and 5 or more units for men).
- Hemax is indicated in non-iron-deficient adults prior to major planned orthopedic surgical procedures with a high risk of transfusion-related complications, to reduce the use of allogeneic blood transfusions. The use of the drug should be limited to adult patients with mild anemia (hemoglobin levels between 10–13 g/dL) who are not participating in an autologous blood collection program, and who are expected to have a moderate blood loss (900–1800 mL).
- Hemax is indicated for the treatment of symptomatic anemia (hemoglobin level ≤ 10 g/dL) in adult patients with low- or intermediate-1-risk primary myelodysplastic syndrome (MDS) and low serum erythropoietin levels (<200 mU/mL).
Contraindications.
- Uncontrolled arterial hypertension;
- development of pure red cell aplasia (PRCA) due to treatment with any erythropoietin (see section "Special precautions");
- hypersensitivity to human albumin;
- hypersensitivity to drugs derived from mammalian cells;
- hypersensitivity to the active substance or any of the excipients of the drug;
- severe coronary, peripheral arterial, carotid, or cerebrovascular diseases, as well as recent myocardial infarction or stroke in patients undergoing major planned orthopedic surgery who have not participated in an autologous blood collection program;
- inability to apply adequate antithrombotic prophylaxis in surgical patients;
- contraindications related to autologous blood collection programs in patients receiving epoetin alfa.
Interaction with other medicinal products and other forms of interaction.
No interactions between Hemax and other medicinal products have been observed.
There are no data indicating that treatment with epoetin alfa affects the metabolism of other drugs.
Medicinal products that suppress erythropoiesis may reduce the response to treatment with epoetin alfa.
Since cyclosporine binds to erythrocytes, a potential drug interaction may occur. When Hemax and cyclosporine are used concomitantly, cyclosporine blood levels should be monitored and the dose adjusted if hematocrit levels increase.
There is no evidence of interaction between epoetin alfa and G-CSF (granulocyte colony-stimulating factor) or GM-CSF (granulocyte-macrophage colony-stimulating factor) regarding hematological differentiation or tumor cell proliferation in in vitro biopsy samples.
In adult women with metastatic breast cancer, subcutaneous administration of epoetin alfa at a dose of 40,000 IU/mL concomitantly with trastuzumab at a dose of 6 mg/kg did not affect the pharmacokinetics of trastuzumab.
Special precautions for use.
Arterial pressure should be continuously monitored in all patients during treatment with Hemax. The drug should be used with caution in patients with untreated hypertension, inadequately treated hypertension, or poorly controlled hypertension. During treatment with Hemax, initiation or intensification of antihypertensive therapy may become necessary. If blood pressure cannot be controlled, administration of epoetin alfa should be discontinued.
Cases of hypertensive crisis with encephalopathy and seizures, requiring immediate medical evaluation and intensive therapy, have also been observed in patients with normal or low blood pressure at the start of treatment. Particular attention should be paid to the sudden onset of a migraine-like, shooting headache.
Epoetin alfa should be used with caution in patients with epilepsy, a history of seizures, or medical conditions that are risk factors for seizures, such as central nervous system (CNS) infections or brain metastases.
Epoetin alfa should be used with caution in patients with chronic hepatic insufficiency. The safety of epoetin alfa in this patient population has not been established.
In patients receiving erythropoiesis-stimulating agents, there is an increased risk of vascular disorders with thrombotic complications, including venous and arterial thrombosis and embolism (including fatal outcomes), such as deep vein thrombosis, pulmonary embolism, retinal vein thrombosis, and myocardial infarction. Cases of stroke (including ischemic stroke, hemorrhagic stroke, and transient ischemic attacks) have also been reported.
Before initiating epoetin alfa therapy, the risks of thrombovascular events with thrombotic complications should be carefully weighed against the expected benefits, especially in patients with existing risk factors, including excessive body weight and a history of thrombovascular events (e.g., deep vein thrombosis, pulmonary embolism, and stroke).
Hemoglobin levels should be closely monitored in all patients due to the potential increased risk of thromboembolic complications and fatal outcomes when the drug is used at hemoglobin levels above the target range indicated for use.
During treatment, a moderate, dose-dependent increase in platelet count within the normal range may occur. This parameter decreases during continued treatment. Cases of thrombocytosis have also been reported. Platelet counts should be monitored regularly during the first 8 weeks of treatment.
Serum ferritin levels (or serum iron concentration) should be determined in all patients before and during treatment with Hemax. Iron supplementation should be administered as needed. Lack of clinical response to Hemax therapy requires investigation for contributing factors such as iron, folic acid, or vitamin B12 deficiency, aluminum intoxication, intercurrent infections, inflammatory processes, trauma, hemolysis, or bone marrow fibrosis of any etiology.
All other causes of anemia (iron, folic acid, or vitamin B12 deficiency, aluminum intoxication, infection or inflammation, blood loss, hemolysis, or bone marrow fibrosis of any origin) must be identified and treated before initiating epoetin alfa therapy or deciding to increase the dose. In most cases, serum ferritin levels decreased simultaneously with an increase in hematocrit.
All patients receiving epoetin alfa should receive adequate iron supplementation (200 mg daily orally) throughout the entire course of therapy. To ensure sufficient iron levels in the body, iron supplementation should be initiated as early as possible, even several weeks before starting an autologous blood donation program.
To ensure an optimal response to epoetin alfa therapy, adequate iron supply must be ensured:
- For patients with chronic kidney disease, iron supplementation (200–300 mg/day for adults and 100–200 mg/day for children orally, calculated as elemental iron) is recommended if serum ferritin levels are below 100 ng/mL;
- For patients with oncological diseases, iron supplementation (200–300 mg/day orally, calculated as elemental iron) is recommended if transferrin saturation is below 20%;
- For patients participating in an autologous blood donation program, iron supplementation (200 mg/day orally, calculated as elemental iron) is recommended several weeks before the start of blood collection to achieve significant iron stores before initiating therapy and during the course of epoetin alfa treatment;
- For patients undergoing extensive elective orthopedic surgery, iron supplementation (200 mg/day orally, calculated as elemental iron) is recommended during the course of epoetin alfa therapy. Iron supplementation should be initiated before starting epoetin alfa therapy, if possible, to achieve significant iron stores.
Very rare cases of development or worsening of existing porphyria have been reported in patients receiving epoetin alfa. Epoetin alfa should be used with caution in patients with porphyria.
Cases of serious, treatment-related skin adverse reactions, including Stevens-Johnson syndrome and toxic epidermal necrolysis, potentially life-threatening or fatal, have been reported with epoetin therapy. More severe cases occurred during treatment with long-acting epoetins.
Patients should be informed about possible skin-related adverse reactions. If symptoms of skin adverse reactions develop, epoetin alfa therapy should be discontinued immediately, and alternative treatment options should be considered.
If a patient develops a severe skin reaction, such as Stevens-Johnson syndrome or toxic epidermal necrolysis, during epoetin therapy, re-administration of the drug is contraindicated.
The trade name of the erythropoiesis-stimulating agent used in treatment must be clearly documented in the patient's medical record. Switching a patient from one erythropoiesis-stimulating agent to another is possible only under physician supervision.
Pure red cell aplasia (PRCA).
Cases of antibody-mediated pure red cell aplasia (PRCA) have been reported after several months or years of subcutaneous epoetin administration, primarily in patients with chronic kidney disease. Cases of pure red cell aplasia have also been reported in patients with hepatitis C receiving interferon and ribavirin concurrently with erythropoiesis-stimulating agents. Epoetin alfa is not indicated for the treatment of anemia associated with hepatitis C.
Patients who experience a sudden loss of therapeutic efficacy (manifested by a hemoglobin decrease of 1–2 g/dL per month) with increased transfusion requirements should be referred for reticulocyte count testing and evaluation for typical causes of reduced clinical response (iron, folic acid, or vitamin B12 deficiency, aluminum intoxication, infection or inflammation, blood loss, hemolysis, or bone marrow fibrosis of any etiology).
In cases of paradoxical hemoglobin decline and development of severe anemia associated with low reticulocyte count, Hemax therapy should be discontinued immediately, and testing for anti-erythropoietin antibodies should be performed, along with bone marrow examination to confirm the diagnosis of pure red cell aplasia (PRCA).
Patients should not be treated with other erythropoiesis-stimulating agents due to the possibility of cross-reactivity.
Treatment of symptomatic anemia in adult and pediatric patients with chronic kidney disease.
In patients with chronic kidney disease receiving epoetin alfa, hemoglobin levels should be monitored regularly until a stable level is achieved, then periodically. The rate of hemoglobin increase should be approximately 1 g/dL (0.62 mmol/L) per month and should not exceed 2 g/dL (1.25 mmol/L) per month to minimize the risk of arterial hypertension.
In patients with chronic kidney disease, the achieved hemoglobin level should not exceed the upper limit of the desired hemoglobin concentration. Clinical studies have shown an increased risk of mortality and serious cardiovascular adverse events when erythropoiesis-stimulating agents are used to achieve hemoglobin concentrations above 12 g/dL (7.5 mmol/L).
Controlled clinical trials have not demonstrated significant benefits of epoetin use at hemoglobin concentrations above the level required to control anemia symptoms and prevent blood transfusions.
The dose of Hemax should be increased cautiously in patients with chronic kidney disease, as high cumulative doses of erythropoietin may be associated with increased risks of mortality, serious cardiovascular, and cerebrovascular events. In patients with inadequate response to epoetin therapy, alternative approaches to manage poor response should be considered.
Patients with chronic kidney disease receiving Hemax subcutaneously should be monitored regularly for loss of treatment efficacy, defined as a decrease or loss of response to epoetin alfa in patients who previously responded to therapy. Loss of efficacy is characterized by a persistent decrease in hemoglobin levels despite increasing the dose of epoetin alfa.
With extended dosing interval regimens (administration of epoetin alfa less than once weekly), hemoglobin levels may decrease in some patients, who may require dose adjustment. Hemoglobin levels should be monitored regularly.
In hemodialysis patients, shunt thrombosis has been observed, particularly in those prone to hypotension or with complications of arteriovenous fistulas (e.g., stenosis, aneurysms, etc.). Such patients should undergo shunt evaluation and receive thrombosis prophylaxis, for example, with acetylsalicylic acid.
Hyperkalemia has been observed in isolated cases, although a causal relationship has not been established. Serum electrolyte levels should be monitored in patients with chronic kidney disease. In case of increased serum potassium levels, in addition to appropriate hyperkalemia treatment, temporary discontinuation of Hemax should be considered until potassium levels normalize.
Due to increased hematocrit, patients on hemodialysis receiving Hemax often require increased heparin doses during dialysis. Inadequate heparinization may lead to dialysis system occlusion.
Based on current information, the use of Hemax for the treatment of anemia in adult pre-dialysis patients with kidney disease does not accelerate the progression of renal failure.
Treatment of patients with anemia due to chemotherapy.
In oncology patients receiving epoetin alfa, hemoglobin levels should be monitored regularly until a stable level is achieved, then periodically.
Epoetins are growth factors that primarily stimulate erythrocyte production. Erythropoietin receptors have also been identified on the surface of various tumor cells. As with other growth factors, stimulation of certain tumor types by epoetins cannot be excluded.
An effect of erythropoiesis-stimulating agents on tumor progression or progression-free survival cannot be ruled out. Studies of epoetin alfa and other erythropoiesis-stimulating agents have been associated with reduced locoregional tumor control or reduced overall survival:
- Reduced locoregional control in patients with progressive head and neck cancer receiving radiotherapy when used to increase hemoglobin levels above 14 g/dL (8.7 mmol/L);
- Reduced overall survival and increased number of deaths due to disease progression within 4 months in patients with metastatic breast cancer receiving chemotherapy when used to increase hemoglobin levels to 12–14 g/dL (7.5–8.7 mmol/L);
- Increased risk of fatal outcomes when used to increase hemoglobin levels to 12 g/dL (7.5 mmol/L) in patients with active malignant disease not receiving chemotherapy or radiotherapy. Erythropoiesis-stimulating agents are contraindicated in this patient group;
- 9% increased risk of disease progression or death in the group of patients receiving epoetin alfa and standard therapy, and a 15% increased risk, which cannot be statistically excluded, in patients with metastatic breast cancer receiving chemotherapy when used to increase hemoglobin levels to 10–12 g/dL (6.2–7.5 mmol/L).
Given the above, in certain clinical situations, blood transfusion may be preferred for the treatment of anemia in cancer patients. The decision to use recombinant erythropoietins should be based on a benefit-risk assessment for the individual patient, considering the specific clinical context. Factors to consider in this assessment should include tumor type and stage; degree of anemia; expected life expectancy; treatment setting; and patient preference.
In oncology patients receiving chemotherapy, a 2–3-week delay between epoetin administration and the appearance of erythropoietin-induced blood cells is typically observed. This should be considered when evaluating the appropriateness of therapy (particularly in patients with transfusion needs).
Patients undergoing surgery and participating in autologous blood donation programs.
All special precautions related to autologous blood donation programs should be observed, particularly procedures for restoring circulating blood volume.
Patients undergoing extensive elective orthopedic surgery.
Standard hemotransfusion practices should always be followed in the pre- and postoperative periods. Patients undergoing extensive elective orthopedic surgery should receive appropriate antithrombotic prophylaxis, as thrombotic and vascular complications may occur after surgery, especially in patients with concomitant cardiovascular diseases. Particular caution should be exercised in treating patients prone to deep vein thrombosis. Moreover, in patients with an initial hemoglobin level > 13 g/dL, the risk of postoperative thrombotic or vascular complications associated with epoetin alfa therapy is significantly higher. Therefore, the use of epoetin alfa in patients with an initial hemoglobin level > 13 g/dL is not recommended.
Diet.
As hematocrit increases, patients' appetite increases, leading to higher food intake. Measures should be taken to prevent increased potassium intake.
Carcinogenic and mutagenic properties.
The carcinogenic properties of Hemax have not been studied. Epoetin does not induce genetic mutations in bacteria or chromosomal aberrations in mammalian cells.
Effect on fertility.
In pregnant rats, a tendency toward a slight increase in embryo mortality was observed after intravenous administration of epoetin at doses of 100–500 IU/kg.
Reduced drug efficacy or lack of response.
If patients receiving epoetin at maintenance doses show reduced response or no response at all, the following causes should be excluded:
- Iron deficiency
- Infection, inflammation, tumors
- Occult blood loss
- Hematological disorders (thalassemia, myelodysplasia, etc.)
- Hemolysis
- Aluminum intoxication
- Vitamin B12 or folic acid deficiency
- Cystic fibrosis
- Pure red cell aplasia.
Sodium chloride.
This medicinal product contains less than 1 mmol (23 mg) of sodium per dose, i.e., it is sodium-free.
Use during pregnancy or breastfeeding.
Pregnancy.
Sufficient studies on the use of Hemax in pregnant women have not been conducted. Animal studies have shown reproductive toxicity. Therefore, this drug should be prescribed to pregnant women only if the potential benefit justifies the potential risk to the fetus. The use of epoetin alfa in pregnant women participating in autologous blood donation programs is not recommended.
In studies in pregnant rats, a slight increase in embryo mortality was observed, whereas in pregnant rabbits receiving the drug at a dose of 500 IU/kg, no adverse effects were observed.
Breastfeeding.
Human erythropoietin is normally present in breast milk, although the role of erythropoietin from breast milk remains unclear. It is unknown whether exogenous epoetin alfa is excreted in breast milk. Epoetin alfa should be used with caution in women who are breastfeeding. The decision to continue or discontinue breastfeeding or to continue or discontinue epoetin alfa therapy should be made, taking into account the benefits of breastfeeding for the child and the benefits of epoetin alfa therapy for the woman.
The use of epoetin alfa in patients participating in autologous blood donation programs during breastfeeding is not recommended.
Fertility.
Studies on the effect of epoetin alfa on fertility in men or women have not been conducted.
Ability to affect reaction speed when driving or operating machinery.
No specific studies have been conducted on this issue, but given that adverse reactions such as headache, fatigue, and others may occur during Hemax use, patients taking this drug should refrain from driving or operating machinery.
Method of Administration and Dosage.
All other causes of anemia (iron deficiency, folic acid or vitamin B12 deficiency, aluminum toxicity, infection or inflammation, blood loss, hemolysis, or bone marrow fibrosis of any etiology) must be identified and treated prior to initiating epoetin alfa therapy, and dosage adjustments should be made if necessary. To achieve an optimal response to epoetin alfa treatment, adequate iron supply must be ensured, and iron supplementation should be administered as needed (see section "Special Precautions").
Treatment of symptomatic anemia in adult patients with chronic kidney disease.
Symptoms and complications of anemia may vary depending on age, sex, and concomitant diseases; therefore, individual patient assessment by a physician is required.
The recommended target hemoglobin level is 10 to 12 g/dL (6.2–7.5 mmol/L). Hemax should be used to increase hemoglobin levels up to a maximum of 12 g/dL (7.5 mmol/L). Increases in hemoglobin exceeding 2 g/dL (1.25 mmol/L) within 4 weeks should be avoided. If such an increase occurs, the dose should be reduced as described below.
Due to individual variability, some patients may achieve hemoglobin levels above or below the desired target range. Hemoglobin levels should be monitored and doses adjusted to maintain levels within the recommended range of 10 g/dL (6.2 mmol/L) to 12 g/dL (7.5 mmol/L).
Sustained hemoglobin levels above 12 g/dL (7.5 mmol/L) should be avoided. If hemoglobin concentration increases by more than 2 g/dL (1.25 mmol/L) per month or sustained levels exceed 12 g/dL (7.5 mmol/L), the Hemax dose should be reduced by 25%. If hemoglobin exceeds 13 g/dL (8.1 mmol/L), treatment should be temporarily discontinued until hemoglobin decreases to 12 g/dL (7.5 mmol/L), after which therapy should be resumed at a dose 25% lower than the previous dose.
Patients should be closely monitored to ensure that the lowest effective dose of Hemax is used to control anemia and its symptoms, maintaining hemoglobin levels no higher than 12 g/dL (7.5 mmol/L).
Dose escalation of erythropoiesis-stimulating agents should be performed cautiously in patients with chronic kidney disease. In patients with poor response to such therapy, other potential causes of inadequate response should be considered (see section "Special Precautions").
Hemax treatment is divided into two phases: the correction phase and the maintenance phase.
Adult patients undergoing hemodialysis.
Intravenous administration is preferred for patients undergoing hemodialysis.
Correction phase.
Initial dose: 50 IU/kg three times weekly.
The dose may be increased or decreased by 25 IU/kg (three times weekly) as needed to achieve the target hemoglobin level of 10–12 g/dL (6.2–7.5 mmol/L). Dose adjustments should be made gradually, no more frequently than once every 4 weeks.
Maintenance phase.
Recommended weekly dose: 75 to 300 IU/kg.
The dose should be adjusted appropriately to maintain the target hemoglobin level of 10 to 12 g/dL (6.2–7.5 mmol/L).
Patients with very low initial hemoglobin levels (< 6 g/dL or < 3.75 mmol/L) may require higher maintenance doses than those with less severe anemia at the start of treatment (hemoglobin > 8 g/dL or > 5 mmol/L).
Adult patients with pre-dialysis chronic kidney disease.
In pre-dialysis patients without an established intravenous catheter, Hemax may be administered subcutaneously.
Correction phase.
Initial dose: 50 IU/kg three times weekly, with dose increases of 25 IU/kg (three times weekly) as needed, at intervals of no less than 4 weeks, until hemoglobin reaches the target range of 10–12 g/dL (6.2–7.5 mmol/L).
Maintenance phase.
During the maintenance phase, Hemax may be administered either three times weekly or, if given subcutaneously, once weekly or once every two weeks.
Doses and administration intervals should be adjusted to maintain the target hemoglobin level of 10 to 12 g/dL (6.2–7.5 mmol/L). Longer intervals between doses may require higher doses.
Maximum dose should not exceed 150 IU/kg three times weekly, 240 IU/kg (maximum up to 20,000 IU) once weekly, or 480 IU/kg (maximum up to 40,000 IU) once every two weeks.
Adult patients undergoing peritoneal dialysis.
In the absence of an established intravenous catheter, Hemax may be administered subcutaneously.
Correction phase.
Initial dose: 50 IU/kg twice weekly.
Maintenance phase.
Recommended maintenance dose: 25 to 50 IU/kg twice weekly, administered as two equal injections.
The dose should be adjusted appropriately to maintain the target hemoglobin level of 10 to 12 g/dL (6.2–7.5 mmol/L).
Treatment of adult patients with anemia due to chemotherapy.
Symptoms and complications of anemia may vary depending on age, sex, and concomitant diseases; therefore, individual assessment by a physician is required.
Hemax should be administered to patients with anemia (hemoglobin ≤ 10 g/dL (6.2 mmol/L)). Initial dose: 150 IU/kg subcutaneously three times weekly.
Alternatively, Hemax may be administered at a dose of 450 IU/kg subcutaneously once weekly.
The dose should be adjusted appropriately to maintain the target hemoglobin level of 10 to 12 g/dL (6.2–7.5 mmol/L). Sustained hemoglobin levels above 12 g/dL (7.5 mmol/L) should be avoided.
If hemoglobin increases by at least 1 g/dL (0.6 mmol/L) or reticulocyte count increases by ≥ 40,000 cells/μL after 4 weeks of treatment, continue Hemax at 150 IU/kg three times weekly or 450 IU/kg once weekly.
If, after 4 weeks of initial dosing, hemoglobin increases by less than 1 g/dL (0.62 mmol/L) or reticulocyte count increases by less than 40,000 cells/μL, the dose should be increased to 300 IU/kg three times weekly.
If, after an additional 4 weeks of treatment at 300 IU/kg three times weekly, hemoglobin increases by 1 g/dL (0.62 mmol/L) or more, or reticulocyte count increases by 40,000 cells/μL or more, continue Hemax at 300 IU/kg three times weekly.
However, if hemoglobin increases by less than 1 g/dL (less than 0.62 mmol/L) or reticulocyte count increases by less than 40,000 cells/μL from baseline, the clinical response is considered inadequate and treatment should be discontinued.
Dose adjustment to maintain target hemoglobin level of 10–12 g/dL.
If hemoglobin increases by more than 2 g/dL (1.25 mmol/L) within one month or exceeds 12 g/dL (7.5 mmol/L), reduce the Hemax dose by 25–50%. If hemoglobin exceeds 13 g/dL (8.1 mmol/L), therapy should be temporarily discontinued until levels fall below 12 g/dL (7.5 mmol/L), then resumed at a dose 25% lower than the previous dose.
Recommended dosing regimen:
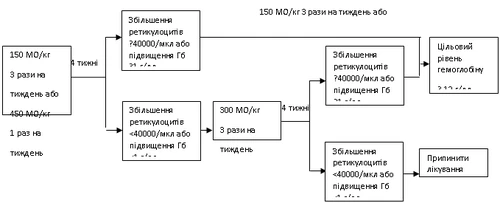
Patients should be closely monitored to ensure that the lowest approved dose of Hemax is used to control anemia symptoms.
Hemax administration should continue for one month after chemotherapy is discontinued.
Adult patients participating in autologous blood donation programs prior to surgery.
Patients with moderate anemia (hematocrit 33–39%) who are expected to require ≥ 4 units of blood should receive Hemax at a dose of 600 IU/kg intravenously twice weekly for 3 weeks prior to surgery. Hemax should be administered after each blood donation procedure.
Adult patients prior to major elective orthopedic surgery.
Recommended dose: 600 IU/kg subcutaneously once weekly for 3 weeks prior to surgery (on days 21, 14, and 7 before surgery) and on the day of surgery.
If medical necessity requires a preoperative period shorter than 3 weeks, Hemax should be administered daily at 300 IU/kg subcutaneously for 10 consecutive days before surgery, on the day of surgery, and for 4 days postoperatively. If hemoglobin reaches 15 g/dL or higher during the preoperative period, Hemax administration must be completely discontinued and no further doses administered.
Adult patients with low- or intermediate-1 risk MDS.
Hemax should be administered to patients with symptomatic anemia (hemoglobin ≤ 10 g/dL (6.2 mmol/L)).
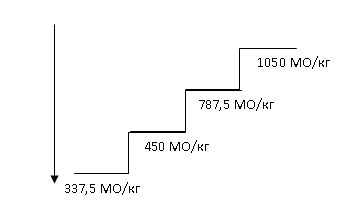
Recommended initial dose: 450 IU/kg (maximum total dose 40,000 IU) subcutaneously once weekly, with at least 5 days between doses.
The dose should be appropriately adjusted to maintain the target hemoglobin level of 10 to 12 g/dL (6.2–7.5 mmol/L). Response to Hemax therapy should be evaluated 8 to 12 weeks after initiation. Dose increases and decreases should be performed stepwise (see diagram below). Hemoglobin levels above 12 g/dL (7.5 mmol/L) should be avoided.
Dose escalation. Dose increases should not exceed 1050 IU/kg (total dose 80,000 IU) per week. If response to treatment is lost or hemoglobin concentration decreases by ≥ 1 g/dL after dose reduction, the dose should be increased by one step. At least 4 weeks should elapse between dose escalations.
Dose maintenance and reduction. Hemax treatment should be temporarily discontinued if hemoglobin concentration exceeds 12 g/dL (7.5 mmol/L). Once hemoglobin falls below 11 g/dL, treatment should be resumed at the same dose or one step lower, at the physician’s discretion. Dose reduction should be considered if hemoglobin rises rapidly (> 2 g/dL within 4 weeks).
 |
|||||
  |
|||||
Symptoms and complications of anemia may vary depending on age, sex, and concomitant diseases; individual assessment of the patient's disease course and condition is required.
Children.
Treatment of symptomatic anemia in children with chronic renal failure undergoing hemodialysis.
Symptoms and complications of anemia may vary depending on age, sex, and concomitant diseases; individual assessment of the patient's disease course and condition is required.
In children, the recommended hemoglobin level is from 9.5 g/dL to 11 g/dL (5.9–6.8 mmol/L). Hemax should be used to increase hemoglobin levels up to, but not exceeding, 11 g/dL (6.8 mmol/L). Increases in hemoglobin levels by more than 2 g/dL (1.25 mmol/L) within 4 weeks should be avoided.
In such cases, the dose should be adjusted appropriately.
Patients should be closely monitored to ensure that the lowest approved dose of Hemax required to control anemia symptoms is used.
Treatment consists of two phases – the correction phase and the maintenance phase.
When an established intravenous catheter is present, the intravenous route of administration is preferred.
Correction phase.
Initial dose: 50 IU/kg intravenously three times weekly.
The dose may be increased or decreased by 25 IU/kg (three times weekly) as needed to achieve the target hemoglobin level of 9.5 g/dL to 11 g/dL (5.9–6.8 mmol/L). Dose adjustments should be made incrementally, no more frequently than once every 4 weeks.
Maintenance phase.
The dose should be adjusted appropriately to maintain the optimal hemoglobin level of 9.5 g/dL to 11 g/dL (5.9–6.8 mmol/L).
Generally, children weighing less than 30 kg require higher maintenance doses than adults and children weighing over 30 kg. Pediatric patients with very low initial hemoglobin levels (< 6.8 g/dL, or < 4.25 mmol/L) may require higher maintenance doses compared to patients with higher initial hemoglobin levels (> 6.8 g/dL, or > 4.25 mmol/L).
Treatment of anemia in children with chronic renal failure during the pre-dialysis period or on peritoneal dialysis.
The safety and efficacy of Hemax in children with chronic renal failure and anemia who are not yet on dialysis or are on peritoneal dialysis have not been established. Currently, no dosage recommendations can be made for this patient group.
Treatment of children with anemia caused by chemotherapy.
The safety and efficacy of Hemax in children with anemia caused by chemotherapy have not been established.
Treatment of children participating in autologous blood donation programs prior to surgical procedures.
The safety and efficacy of Hemax in children participating in autologous blood donation programs prior to major surgical procedures have not been established.
Treatment of children prior to elective orthopedic surgery.
The safety and efficacy of Hemax in children undergoing elective orthopedic surgery have not been established.
Administration method
Epoetin alfa can be administered via subcutaneous or intravenous injection.
Preparation of the injection solution: Add the volume of water for injection specified in the table below to each vial containing the lyophilisate.
| Active ingredient |
1000 IU |
2000 IU |
3000 IU |
4000 IU |
10 000 IU |
20 000 IU |
40 000 IU |
| Water for injections |
1 ml |
2 ml |
2 ml |
2 ml |
1 ml |
1 ml |
1 ml |
As with any parenteral medicinal product, the epoetin alfa solution should be inspected visually for particulate matter and discoloration prior to administration.
Intravenous administration.
Hemax is administered intravenously to adult patients participating in an autologous blood donation program prior to surgical procedures. For treatment of symptomatic anemia in adults and children with chronic renal failure who have intravenous access (hemodialysis patients), the intravenous route is preferred.
Hemax is administered by intravenous injection over a period of 1 to 5 minutes, depending on the dose. For patients undergoing hemodialysis, the bolus injection may be given directly during the procedure via an appropriate venous port in the dialysis line. Alternatively, the drug may be administered after completion of hemodialysis through the fistula or catheter, followed by administration of 10 mL of isotonic sodium chloride solution to flush the system and ensure proper distribution of the drug in the circulation.
Slow administration is recommended for patients who experience influenza-like symptoms during treatment (see section "Adverse reactions").
Hemax must not be administered by intravenous infusion or mixed with other medicinal products.
Subcutaneous administration.
Hemax is administered subcutaneously to adult patients with anemia due to chemotherapy, to patients prior to elective major orthopedic surgery, and to adult patients with low- or intermediate-1 risk MDS.
For treatment of symptomatic anemia in adult patients with chronic renal failure during the pre-dialysis period or on peritoneal dialysis, in the absence of an established intravenous catheter, subcutaneous administration of Hemax may be used.
The maximum volume for subcutaneous injection at a single site is 1 mL. If larger volumes are required, subcutaneous injections should be divided among multiple sites.
The subcutaneous injection should be administered in the extremities or the anterior abdominal wall.
If, in the physician’s opinion, the patient or caregiver is capable of safely and effectively self-administering Hemax subcutaneously, they should be properly instructed in the correct dosage and administration technique.
Children.
Epoetin alfa is indicated for the treatment of anemia associated with chronic renal failure in children aged 1 year to 18 years who are on dialysis. The safety and efficacy of the drug in children under 1 month of age have not been established.
Overdose.
The drug has a wide therapeutic window. Overdose of epoetin alfa results in effects reflecting the maximal pharmacological action of the hormone, i.e., polycythemia and associated symptoms such as headache, dizziness, somnolence, etc. In cases of excessively high hemoglobin levels, phlebotomy may be performed. Symptomatic treatment should be administered if necessary.
In case of overdose, the patient should seek immediate medical attention at the nearest healthcare facility or a toxicology center.
Side effects
The most common adverse reaction during treatment with epoetin alfa is dose-dependent elevation of blood pressure or worsening of pre-existing hypertension. Blood pressure monitoring should be initiated from the beginning of treatment. Other common adverse reactions observed during clinical trials with epoetin alfa include deep vein thrombosis, pulmonary embolism, seizures, diarrhea, nausea, headache, flu-like symptoms, pyrexia, rash, and vomiting.
At the beginning of treatment, symptoms resembling a cold may occur, such as headache, muscle and joint pain, and chills. The frequency may vary depending on the indication.
During studies evaluating extended dosing intervals of the drug in adult patients with chronic kidney disease in the pre-dialysis period, worsening of airway patency, including upper airways, nasal congestion, and nasopharyngitis were observed.
An increased incidence of thrombovascular complications has been observed in patients receiving erythropoiesis-stimulating agents (see section "Special precautions").
Albumin (human)
Hemax contains albumin derived from human blood. The risk of transmission of viral infections is extremely low due to the manufacturing process used for this component. The theoretical risk of transmission of the agent causing Creutzfeldt-Jakob disease is also extremely low. There have been no reported cases of transmission of viral agents associated with albumin.
Frequency of adverse reactions: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1000 to <1/100); rare (≥1/10,000 to <1/1000); very rare (<1/10,000); frequency not known (cannot be estimated from available data).
Blood and lymphatic system disorders
Rare – thrombocytosis, pure red cell aplasia.
Immune system disorders
Uncommon – hypersensitivity reactions.
Rare – anaphylactic reactions: potentially serious complications associated with respiratory distress or hypotension; immune responses (has minimal capacity to induce antibody formation).
Nervous system disorders
Common – headache.
Uncommon – seizures, stroke, cerebral hemorrhage.
Frequency not known – cerebrovascular stroke, hypertensive encephalopathy, transient ischemic attack, dizziness, somnolence.
Eye disorders
Frequency not known – retinal vein thrombosis.
Cardiac disorders
Frequency not known – myocardial infarction.
Vascular disorders
Common – venous and arterial thrombosis, arterial hypertension.
Frequency not known – deep vein thrombosis (in patients with chronic kidney disease), hypertensive crisis.
Respiratory, thoracic and mediastinal disorders
Common – pulmonary embolism (in cancer patients), cough.
Uncommon – worsening of airway patency.
Frequency not known – pulmonary embolism (in patients with chronic kidney disease).
Gastrointestinal disorders
Very common – nausea, diarrhea, vomiting.
Skin and subcutaneous tissue disorders
Common – rash, eczema.
Uncommon – urticaria.
Frequency not known – angioneurotic edema, pruritus, Quincke's edema, Stevens-Johnson syndrome, toxic epidermal necrolysis (which may be life-threatening or fatal).
Musculoskeletal, connective tissue and bone disorders
Common – arthralgia, bone pain, limb pain, myalgia.
Congenital, familial and genetic disorders
Rare – acute porphyria.
General disorders and administration site conditions
Very common – fever, pyrexia (in cancer patients).
Common – flu-like symptoms, chills, injection site reactions, peripheral edema.
Frequency not known – chills, lack of response to treatment.
Investigations
Rare – presence of antibodies to erythropoietin.
Frequency not known – hyperphosphatemia, increased plasma concentrations of urea, creatinine, and uric acid (in patients with chronic kidney disease).
Metabolism and nutrition disorders
Uncommon – hyperkalemia (common in patients undergoing hemodialysis).
Injury, poisoning and procedural complications
Common – shunt thrombosis, including dialysis equipment (in patients with chronic kidney disease).
Patients with chronic kidney disease
In patients with chronic kidney disease, hemoglobin levels above 12 g/dL may be associated with an increased risk of cardiovascular complications, including fatal outcomes.
Shunt thrombosis has been reported in patients undergoing hemodialysis, particularly in those with a predisposition to hypotension or complications related to arteriovenous fistula (e.g., stenosis, aneurysms, etc.).
Patients with oncological diseases
Thrombotic complications may occur in patients receiving erythropoiesis-stimulating agents, including epoetin alfa.
Surgical patients
It cannot be excluded that treatment with epoetin alfa in patients with stable hemoglobin levels >13 g/dL may be associated with an increased risk of postoperative thrombotic/vascular complications.
Description of selected adverse reactions
Venous and arterial thromboses, with and without fatal outcome, such as deep vein thrombosis, pulmonary embolism, retinal thrombosis, arterial thrombosis (including myocardial infarction and myocardial ischemia), retinal thrombosis, shunt thrombosis (including occlusion of dialysis system), thromboses at the site of arteriovenous anastomosis. Cerebrovascular complications (including ischemic stroke, cerebral hemorrhage) and transient ischemic attacks, aneurysms may also occur.
Hypersensitivity reactions have been reported, including rash (including urticaria), anaphylactic reactions, and angioneurotic edema.
Severe skin-related adverse reactions associated with epoetin treatment, including Stevens-Johnson syndrome and toxic epidermal necrolysis, which may be life-threatening or fatal, have been reported (see section "Special precautions").
Cases of hypertensive crisis with encephalopathy and seizures requiring immediate medical evaluation and intensive therapy have been observed in patients with normal or low blood pressure at the start of treatment. Particular attention should be paid to the sudden onset of severe, shooting, migraine-like headache, which may be a warning sign.
Pure red cell aplasia
Since epoetin alfa is a protein-based drug, antibody formation may occur in some patients. Rare cases of pure red cell aplasia occurring in patients with kidney failure receiving subcutaneous administration of the drug are associated with the presence of neutralizing antibodies to epoetin alfa. In such cases, the use of any erythropoietin-containing product is contraindicated.
Very rare cases (<1/10,000 cases per patient-year) of antibody-mediated pure red cell aplasia (PRCA) have been reported in patients treated with erythropoietin products for months or years.
Adult patients with low or intermediate-1 risk MDS
In a clinical trial, thrombovascular events (sudden death, ischemic stroke, embolism, phlebitis) occurred in 4 (4.7%) patients. All thrombovascular events occurred in the epoetin alfa group and within the first 24 weeks of treatment. Three cases were confirmed; the fourth case (sudden death) was not confirmed. Two patients had significant risk factors (atrial fibrillation, heart failure, thrombophlebitis).
Children with chronic kidney disease on hemodialysis
Experience with the use of epoetin in children with chronic kidney disease on hemodialysis during clinical trials and in the post-marketing period is limited. No adverse reactions specific to pediatric patients have been identified, nor have any adverse reactions been observed that are not consistent with the underlying disease.
Reporting of suspected adverse reactions after drug authorization is important. It allows continuous monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals are encouraged to report any suspected adverse reactions via the national reporting system.
Shelf life. 2 years.
Storage conditions.
Store out of reach of children, at a temperature not exceeding 25 °C.
Packaging.
1 vial per cardboard box.
Prescription status. Prescription only.
Manufacturer. Biosidus S.A. /
Biosidus S.A.
Manufacturer's address and place of business.
Constitucion 4234 (zip code C1254ABX), City of Buenos Aires, Argentine Republic /
Constitucion 4234 (zip code C1254ABX), of the City of Buenos Aires, Argentine Republic.