Advait
UkraineTable of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT ADYNOVATE (ADVATE)
Composition:
Active substance: coagulation factor VIII (octocog alfa);
1 vial contains:
250 IU* of recombinant human blood coagulation factor VIII (rDNA)**, octocog alfa, which after reconstitution yields approximately 50 IU/mL of recombinant human blood coagulation factor VIII, octocog alfa;
500 IU* of recombinant human blood coagulation factor VIII (rDNA)**, octocog alfa, which after reconstitution yields approximately 100 IU/mL of recombinant human blood coagulation factor VIII, octocog alfa;
1000 IU* of recombinant human blood coagulation factor VIII (rDNA)**, octocog alfa, which after reconstitution yields approximately 200 IU/mL of recombinant human blood coagulation factor VIII, octocog alfa;
1500 IU* of recombinant human blood coagulation factor VIII (rDNA)**, octocog alfa, which after reconstitution yields approximately 300 IU/mL of recombinant human blood coagulation factor VIII, octocog alfa;
2000 IU* of recombinant human blood coagulation factor VIII (rDNA)**, octocog alfa, which after reconstitution yields approximately 400 IU/mL of recombinant human blood coagulation factor VIII, octocog alfa;
3000 IU* of recombinant human blood coagulation factor VIII (rDNA)**, octocog alfa, which after reconstitution yields approximately 600 IU/mL of recombinant human blood coagulation factor VIII, octocog alfa;
Excipients: trehalose, histidine, tris(hydroxymethyl)aminomethane, sodium chloride, calcium chloride, glutathione (reduced), polysorbate 80, mannitol.
* Activity (in international units) is determined using a chromogenic quantitative assay relative to an internal standard calibrated against the WHO standard. Specific activity is approximately 4000 – 10000 IU/mg protein.
** Recombinant human coagulation factor VIII is produced using recombinant DNA technology in Chinese hamster ovary cells. No human- or animal-derived proteins (exogenous) are added during cell culture, purification, or final formulation steps.
Pharmaceutical form. Powder and solvent for solution for injection.
Main physicochemical properties: friable white or almost white powder; after reconstitution – clear, colourless solution free from foreign particles.
Pharmacotherapeutic group. Blood and blood-forming organs. Antihemorrhagics. Vitamin K and other hemostatic agents. Coagulation factors. Coagulation factor VIII.
ATC code B02BD02.
Pharmacological properties.
Pharmacodynamics.
The factor VIII/von Willebrand factor complex consists of two molecules (factor VIII and von Willebrand factor) with different physiological functions. ADVATE contains recombinant human coagulation factor VIII (octocog alfa), a glycoprotein that is biologically equivalent to the factor VIII glycoprotein in human plasma.
Octocog alfa is a glycoprotein composed of 2332 amino acids with an approximate molecular weight of 280 kDa. When infused into a patient with haemophilia, octocog alfa binds to endogenous von Willebrand factor in the patient's circulatory system. Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X to activated factor X. Activated factor X then converts prothrombin to thrombin. Thrombin subsequently converts fibrinogen into fibrin, leading to the formation of a fibrin clot. Haemophilia A is an X-linked inherited bleeding disorder caused by reduced levels of factor VIII activity, resulting in prolonged bleeding into joints, muscles, or internal organs, occurring spontaneously or following trauma or surgery. Replacement therapy increases plasma factor VIII levels, enabling temporary correction of factor VIII deficiency and reducing bleeding frequency and tendency.
Data on immune tolerance induction (ITI) in patients with inhibitors to factor VIII have been collected. In sub-study 060103 involving previously untreated children, ITI procedures were documented in 11 such patients. A retrospective analysis was performed on 30 paediatric patients undergoing ITI (in study 060703). In a prospective non-interventional registry (PASS-INT-004), ITI was documented in 44 paediatric and adult patients, 36 of whom completed ITI therapy. The data indicate that immune tolerance can be achieved.
In study 060201, 53 previously treated patients were enrolled to compare two long-term prophylactic treatment regimens: an individual pharmacokinetic-based dosing regimen (20–80 IU factor VIII per kg body weight at intervals of 72 ± 6 hours, n = 23) versus a standard prophylactic dosing regimen (20–40 IU/kg every 48 ± 6 hours, n = 30). The pharmacokinetic-based regimen was designed to maintain minimum factor VIII levels ≥ 1% throughout a 72-hour dosing interval. Data from this study indicate that both prophylactic regimens are comparable in terms of reducing bleeding frequency.
The European Medicines Agency has waived the obligation to submit results of studies with ADVATE in all paediatric subgroups of patients with haemophilia A (congenital factor VIII deficiency) for the following indications: "Immune tolerance induction (ITI) in patients with haemophilia A (congenital factor VIII deficiency) who have developed factor VIII inhibitors" and "Treatment and prevention of bleeding in patients with haemophilia A (congenital factor VIII deficiency)" (see section "Posology and method of administration" for information on use in paediatric patients).
Pharmacokinetics.
All pharmacokinetic studies with ADVATE were conducted in previously treated patients with severe and moderate haemophilia A (baseline factor VIII levels ≤ 2%). Plasma samples were analysed at a central laboratory using a one-stage assay.
Overall, pharmacokinetic parameters from 195 patients with severe haemophilia A (baseline factor VIII < 1%) were included in the per-protocol pharmacokinetic analysis dataset. Categories used for summarizing pharmacokinetic parameters were infants (aged 1 month to < 2 years), young children (aged 2 to < 5 years), older children (aged 5 to < 12 years), adolescents (aged 12 to < 18 years), and adults (aged 18 years and older), with age defined as the age at the time of the PK infusion.
Table 1
| Summary of pharmacokinetic parameters of ADYNOVI in patients with severe haemophilia A (baseline factor VIII < 1%) |
|||||
| Parameter (mean ± standard deviation) |
Infants (n = 5) |
Young children (n = 30) |
Older children (n = 18) |
Adolescents (n = 33) |
Adults (n = 109) |
| Total AUC (IU·hr/dL) |
1362.1 ± 311.8 |
1180.0 ± 432.7 |
1506.6 ± 530.0 |
1317.1 ± 438.6 |
1538.5 ± 519.1 |
| Adjusted incremental recovery at Cmax (IU/dL per IU/kg)a |
2.2 ± 0.6 |
1.8 ± 0.4 |
2.0 ± 0.5 |
2.1 ± 0.6 |
2.2 ± 0.6 |
| Half-life (hr) |
9.0 ± 1.5 |
9.6 ± 1.7 |
11.8 ± 3.8 |
12.1 ± 3.2 |
12.9 ± 4.3 |
| Maximum plasma concentration after infusion (IU/dL) |
110.5 ± 30.2 |
90.8 ± 19.1 |
100.5 ± 25.6 |
107.6 ± 27.6 |
111.3 ± 27.1 |
| Elimination half-life (hr) |
11.0 ± 2.8 |
12.0 ± 2.7 |
15.1 ± 4.7 |
15.0 ± 5.0 |
16.2 ± 6.1 |
| Volume of distribution at steady state (L/kg) |
0.4 ± 0.1 |
0.5 ± 0.1 |
0.5 ± 0.2 |
0.6 ± 0.2 |
0.5 ± 0.2 |
| Clearance (mL/kg·hr) |
3.9 ± 0.9 |
4.8 ± 1.5 |
3.8 ± 1.5 |
4.1 ± 1.0 |
3.6 ± 1.2 |
a Calculated as Cmax – baseline factor VIII level, divided by the dose in IU/kg, where Cmax is the peak post-infusion factor VIII level.
The safety and haemostatic efficacy of ADYNOVATE in the paediatric population are comparable to those in adult patients. Adjusted recovery and terminal half-life (t½) were approximately 20% lower in younger children (up to 6 years) than in adults, which may be partially due to the known increase in plasma volume per kilogram body weight in younger patients.
Currently, there are no pharmacokinetic data available for ADYNOVATE in previously untreated patients.
Clinical characteristics.
Indications.
Treatment and prevention of bleeding episodes in patients with haemophilia A (congenital factor VIII deficiency). ADYNOVATE is indicated for patients of all age groups.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients listed in the "Composition" section, or to mouse or hamster proteins.
Interaction with other medicinal products and other forms of interaction.
Interaction studies for ADYNOVATE have not been conducted.
Special precautions for use.
Traceability
In order to improve traceability of biological medicinal products, the name and batch number of the administered product should be recorded.
Hypersensitivity
Hypersensitivity reactions, including anaphylaxis, have been reported with the use of ADYVATE. The product contains trace amounts of mouse and hamster proteins. Patients should be advised to immediately discontinue administration and seek medical attention if symptoms of hypersensitivity occur. Patients should be informed about early signs of hypersensitivity reactions, such as urticaria, generalized urticaria, chest tightness, wheezing, hypotension, and anaphylaxis.
In case of shock, standard anti-shock measures should be initiated.
Inhibitors
Development of neutralizing antibodies (inhibitors) to factor VIII is a known complication in the treatment of patients with haemophilia A. These inhibitors are usually immunoglobulins IgG directed against the procoagulant activity of factor VIII, measured in Bethesda Units (BU) per 1 ml of plasma using a modified assay. The risk of inhibitor development depends on disease severity and the degree of factor VIII exposure, and is highest during the first 20 exposure days. In rare cases, inhibitors may develop after more than 100 exposure days.
Cases of recurrence of inhibitor development (with low titre) have been observed more than 100 days after switching from one recombinant factor VIII product to another in previously treated patients with a history of inhibitor development. Therefore, careful monitoring for inhibitor development is required after changing factor VIII products.
The clinical significance of inhibitor formation depends on its titre. Low-titre inhibitors, which are transient or remain persistently low, pose a lower risk of inadequate clinical response compared to high-titre inhibitors.
All patients receiving factor VIII therapy should be closely monitored for inhibitor development through clinical observation and laboratory testing. If expected plasma factor VIII activity levels are not achieved or if bleeding is not controlled with an appropriate dose, testing for the presence of factor VIII inhibitors should be performed. In patients with high inhibitor titres, factor VIII therapy may be ineffective, and alternative treatment options should be considered. Such patients should be managed by physicians experienced in the treatment of haemophilia and factor VIII inhibitor management.
Complications related to catheterization
If a central venous access device (CVAD) is required, there is a risk of CVAD-related complications, including local infections, bacteraemia, and thrombosis at the catheter site.
Factors related to excipients
Sodium
This medicinal product contains 10 mg of sodium per vial, equivalent to 0.5% of the WHO recommended maximum daily intake of 2 g sodium for an adult.
It is strongly recommended to record the name and batch number of the medicinal product each time ADYVATE is administered to a patient, in order to establish a link between the patient and the product batch.
Children
Warnings and safety precautions in children do not differ from those in adults.
Use during pregnancy and breastfeeding.
Reproductive toxicity studies of factor VIII in animals have not been conducted. Due to the low prevalence of haemophilia A in women, there is limited experience with the use of factor VIII during pregnancy and breastfeeding. Therefore, factor VIII should be used during pregnancy and breastfeeding only if clearly indicated.
Ability to affect reaction rate when driving or operating machinery.
ADYVATE has no influence on the ability to drive or operate machinery.
Method of Administration and Dosage
Treatment should be initiated under the supervision of a physician experienced in the management of hemophilia; resuscitation equipment should also be available in case of anaphylaxis.
Dosage
The dosage and duration of replacement therapy depend on the severity of factor VIII deficiency, the location and extent of bleeding, as well as the patient's clinical condition.
The amount of factor VIII units is expressed in international units (IU), which are related to the WHO standard for factor VIII preparations. Factor VIII activity in plasma is expressed either as a percentage (relative to normal human plasma) or in IU (relative to the international standard for factor VIII in plasma).
1 international unit (IU) of factor VIII activity is equivalent to the amount of factor VIII present in 1 ml of normal human plasma.
On-demand treatment
The required dose of factor VIII is calculated based on the empirically established relationship: 1 IU of factor VIII per kg of body weight increases factor VIII activity by 2 IU/dL in plasma. The required dose is determined by the following formula:
Required dose (IU) = body weight (kg) × desired increase in factor VIII level (%) × 0.5
In the event of ongoing bleeding episodes, factor VIII activity should not fall below certain plasma activity levels (expressed as % of normal or IU/dL) during the appropriate period. The table below (Table 2) can be used as a guide for dose selection in bleeding episodes and surgical procedures.
Table 2
| Guidance for Dose Selection in Bleeding and Surgical Procedures |
||
| Severity of Bleeding / Type of Surgical Procedure |
Required Factor VIII Level (% or IU/dL) |
Dosing Frequency (hours) / Duration of Therapy (days) |
| Bleeding |
||
| Early hemarthrosis, muscle bleeding, or oral cavity bleeding. |
20–40 |
Repeat injections every 12–24 hours (every 8–24 hours in patients under 6 years of age) for at least 1 day, until bleeding stops as indicated by relief of pain or until healing is achieved. |
| Advanced hemarthrosis, muscle bleeding, or hematoma. |
30–60 |
Repeat injections every 12–24 hours (every 8–24 hours in patients under 6 years of age) for 3–4 days or longer, until pain and disability are resolved. |
| Life-threatening bleeding. |
60–100 |
Repeat injections every 8–24 hours (every 6–12 hours in patients under 6 years of age) until the life-threatening condition is resolved. |
| Surgical Procedure |
||
| Minor, including dental extractions |
30–60 |
Every 24 hours (every 12–24 hours in patients under 6 years of age) for no less than 1 day, until healing is achieved. |
| Major |
80–100 (pre- and post-operatively) |
Repeat injections every 8–24 hours (every 6–24 hours in patients under 6 years of age) until adequate healing; continue treatment for at least 7 additional days to maintain Factor VIII activity between 30% and 60% (IU/dL). |
The dose and frequency of administration should be adjusted according to the clinical situation in each individual case. Under certain circumstances (e.g., presence of an inhibitor with low titer), a higher dose than that calculated by the formula may be required.
During treatment, it is advisable to monitor plasma factor VIII activity levels and use these values to determine the required dose and frequency of repeat injections. In the case of major surgical procedures, precise monitoring of replacement therapy using quantitative factor VIII activity assays in plasma is essential. Different patients may exhibit variable responses to factor VIII, achieving different levels of in vivo recovery and demonstrating different elimination half-lives.
Prophylaxis
For long-term prevention of bleeding episodes in patients with severe hemophilia A, usual doses are 20–40 IU of factor VIII per kg body weight, administered every 2 to 3 days.
Route of administration
ADYVATE should be administered intravenously. If administered by a non-medical professional, appropriate training is required.
The infusion rate should be adjusted to a comfortable level for the patient and must not exceed 10 mL/minute.
After reconstitution, the solution should be clear, colorless, free of foreign particles, and have a pH between 6.7 and 7.3.
Instructions for reconstitution prior to administration
ADYVATE should be administered intravenously after reconstitution.
Visually inspect the reconstituted solution for the presence of particulate matter and/or discoloration.
After reconstitution, the solution should be clear, colorless, and free of particulate matter. Do not use solutions that are cloudy or contain particles.
- A syringe with a Luer-lock connector should be used for administration.
- Use within 3 hours after reconstitution.
- Do not refrigerate the reconstituted solution.
Any unused product or waste material should be disposed of in accordance with local requirements.
Reconstitution using the BAXJECT II device
- Only use sterile water for injection and the diluent device provided in the package for reconstitution.
- Do not use if the BAXJECT II device, its sterile barrier system, or packaging are damaged or show signs of deterioration.
- Aseptic techniques must be followed.
- If the product has been stored in the refrigerator, remove both vials – the ADYVATE powder vial and the diluent vial – and allow them to reach room temperature (approximately 15–25 °C).
- Wash hands thoroughly with warm water and soap.
- Remove the caps from the powder and diluent vials.
- Clean the stoppers with alcohol swabs. Place the vials on a clean, flat surface.
- Open the BAXJECT II device package by removing the paper lid, taking care not to touch the device inside (Fig. a). Do not remove the device from the package. Do not use if the BAXJECT II device, its sterile barrier system, or packaging are damaged or show signs of deterioration.
- Turn the package upside down and insert the clear plastic spike into the diluent vial stopper. Hold the package by the edge and remove it from the BAXJECT II device (Fig. b). Do not remove the blue cap from the BAXJECT II device.
- For reconstitution, use only the sterile water for injection and the diluent device provided in the package. Turn the system (BAXJECT II device attached to the diluent vial) upside down so that the diluent vial is above the device. Insert the white plastic spike into the stopper of the ADYVATE vial. The vacuum will draw the diluent into the ADYVATE vial (Fig. c).
- Gently swirl until the product is completely dissolved. Ensure that ADYVATE is fully dissolved; otherwise, not all of the reconstituted solution may pass through the device filter. The product dissolves rapidly (usually within less than 1 minute). After reconstitution, the solution should be clear, colorless, and free of particulate matter.
Fig. a Fig. b Fig. c
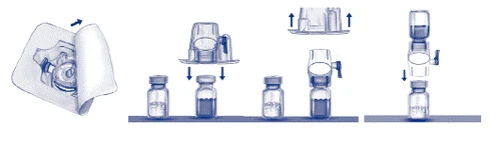
Administration
Follow aseptic techniques.
Parenteral medicinal products should be visually inspected for particulate matter immediately before use, if the solution and container permit. Only clear, colorless solutions should be used.
- Remove the blue cap from the BAXJECT II. Do not draw air into the syringe. Attach the syringe to the BAXJECT II.
- Invert the system (the vial with reconstituted solution should be on top). Slowly draw the reconstituted solution into the syringe by pulling back the plunger.
- Detach the syringe.
- Attach a butterfly needle to the syringe. Administer intravenously. The solution should be infused slowly at a rate comfortable for the patient, not exceeding 10 mL per minute. Pulse rate should be monitored before and during administration of ADYVATE. If a significant increase in pulse rate occurs, reduce the infusion rate or temporarily interrupt the injection, which is usually sufficient to rapidly resolve symptoms (see sections "Special precautions" and "Adverse reactions").
From a microbiological standpoint, the product should be used immediately after reconstitution. However, chemical and physical stability of the medicinal product after opening has been demonstrated for 3 hours at 25 °C.
During its shelf life, the product may be stored at room temperature (not exceeding 25 °C) for one period not exceeding 6 months. The expiration date after 6 months of storage at room temperature must be indicated on the medicinal product packaging. The product must not be returned to the refrigerator for further storage.
Children
For on-demand treatment, dosing in pediatric patients (0 to 18 years of age) does not differ from dosing in adults. For prophylactic therapy in patients under 6 years of age, 20–50 IU of factor VIII per kg body weight should be administered 3–4 times per week.
Overdose.
There are no reports of symptoms associated with overdose of this medicinal product.
Adverse Reactions
Clinical trials of the medicinal product ADYNOVATE included 418 studies with single-dose administration of ADYNOVATE, reporting 93 adverse reactions. The most common adverse reactions included development of neutralizing antibodies to factor VIII (inhibitors), headache, and fever.
Hypersensitivity or allergic reactions (which may include angioedema, burning and stinging at the infusion site, chills, flushing, generalized urticaria, headache, hives, hypotension, lethargy, nausea, restlessness, tachycardia, chest tightness, tingling, vomiting, wheezing) were observed rarely, and in some cases progressed to acute anaphylaxis (including shock).
Hypersensitivity reactions may occur in patients who develop antibodies to mouse and/or hamster proteins.
In patients with haemophilia A receiving factor VIII, including ADYNOVATE, neutralizing antibodies (inhibitors) may develop. If such inhibitors occur, the clinical response may be insufficient. In these cases, it is recommended to consult a specialized haemophilia treatment centre.
List of adverse reactions in tabular form
Table 3 below lists adverse reactions reported during clinical trials and spontaneous reports, categorized by frequency of occurrence. The table follows the MedDRA system organ classes (SOCs) and preferred terms.
Frequency categories are defined as follows: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000), and not known (cannot be estimated from available data). Within each frequency category, adverse reactions are listed in order of decreasing severity.
Table 3
Frequency of adverse reactions reported in clinical trials and spontaneous reports
| Standard MedDRA System Organ Class |
Adverse Reactions |
Frequencya |
| Infections and parasitic infections |
Influenza |
Uncommon |
| Laryngitis |
Uncommon |
|
| Blood and lymphatic system disorders |
Inhibition of factor VIII |
Uncommon (POP)d Very common (PBL)d |
| Lymphangitis |
Uncommon |
|
| Immune system disorders |
Anaphylactic reaction |
Not known |
| Hypersensitivityc |
Not known |
|
| Nervous system disorders |
Headache |
Common |
| Dizziness |
Uncommon |
|
| Memory impairment |
Uncommon |
|
| Syncope |
Uncommon |
|
| Tremor |
Uncommon |
|
| Migraine |
Uncommon |
|
| Dysgeusia |
Uncommon |
|
| Eye disorders |
Eye inflammation |
Uncommon |
| Cardiac disorders |
Palpitations |
Uncommon |
| Vascular disorders |
Haematoma |
Uncommon |
| Flushing |
Uncommon |
|
| Pallor |
Uncommon |
|
| Respiratory, thoracic and mediastinal disorders |
Dyspnoea |
Uncommon |
| Gastrointestinal disorders |
Diarrhoea |
Uncommon |
| Upper abdominal pain |
Uncommon |
|
| Nausea |
Uncommon |
|
| Vomiting |
Uncommon |
|
| Skin and subcutaneous tissue disorders |
Pruritus |
Uncommon |
| Rash |
Uncommon |
|
| Hyperhidrosis |
Uncommon |
|
| Urticaria |
Uncommon |
|
| General disorders and administration site conditions |
Hypothermia |
Common |
| Peripheral oedema |
Uncommon |
|
| Chest pain |
Uncommon |
|
| Chest discomfort |
Uncommon |
|
| Chills |
Uncommon |
|
| Malaise |
Uncommon |
|
| Vascular puncture site haematoma |
Uncommon |
|
| Fatigue |
Not known |
|
| Injection site reaction |
Not known |
|
| Feeling unwell |
Not known |
|
| Investigations |
Increased monocyte count |
Uncommon |
| Decreased blood coagulation factor VIII levelb |
Uncommon |
|
| Decreased haematocrit |
Uncommon |
|
| Investigations abnormal |
Uncommon |
|
| Injury, poisoning and procedural complications |
Post-procedural complications |
Uncommon |
| Post-procedural haemorrhage |
Uncommon |
|
| Procedural site reaction |
Uncommon |
a Calculated based on the total number of patients who received ADYNOVATE (418).
b In one patient, an unexpected decrease in factor VIII coagulation levels occurred during continuous infusion of ADYNOVATE after surgery (10–14 days post-operatively). Haemostasis was maintained throughout this period, and factor VIII plasma levels and clearance rate returned to normal by day 15 post-operatively. Quantitative assays for factor VIII inhibitors performed after completion of continuous infusion and after study completion were negative.
c Explanation regarding this adverse reaction is provided in the section below.
d Frequency is based on data from studies with all factor VIII coagulation products that included patients with severe haemophilia A. PUP – previously untreated patients; PTP – previously treated patients.
Description of selected adverse reactions
Adverse reactions to process residuals
Among 229 patients receiving treatment, antibodies to Chinese hamster ovary (CHO) cell proteins were assessed: 3 patients showed statistically significant trends in titres, 4 patients showed sustained peaks or transient peak potential, and 1 patient showed both findings, but without any clinical symptoms. Among the 229 patients tested for antibodies to mouse IgG, 10 showed statistically significant increases, 2 showed sustained peaks or transient peak potential, and 1 patient showed both findings. Four patients reported isolated events of urticaria, pruritus, rash, and slightly elevated eosinophil counts upon repeated administration of the medicinal product.
Hypersensitivity
Allergic-type reactions include anaphylaxis and may present as dizziness, paraesthesia, rash, flushing, facial swelling, urticaria, and pruritus.
Paediatric
Apart from the development of neutralising antibodies (inhibitors) in previously untreated children and complications related to catheterisation, no differences in adverse reactions were observed in clinical studies.
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after medicinal product authorisation is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare and pharmaceutical professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the automated pharmacovigilance information system at: https://aisf.dec.gov.ua.
Shelf life.
2 years.
Diluent (water for injection) – 5 years.
Storage conditions.
Store at 2 to 8 °C. Do not freeze! Store in the original packaging to protect from light.
Keep out of the reach of children.
Incompatibilities
Since compatibility studies have not been conducted, this medicinal product must not be mixed with other medicinal products or solvents.
Packaging.
One vial of powder for solution for injection (250 IU, 500 IU, 1000 IU, 1500 IU, 2000 IU, or 3000 IU) with one vial of diluent (5 ml water for injection) and one BAXJECT II reconstitution device per carton.
Prescription category.
Prescription only.
Manufacturer.
Baxalta Belgium Manufacturing SA, Belgium / Baxalta Belgium Manufacturing SA, Belgium.
Manufacturer's location and address of place of business.
Boulevard Rene Branquart 80, Lessines, 7860, Belgium / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.