Rixubis
UkrainaSpis treści
INSTRUKCJA stosowania leku RIXUBIS
Skład:
substancja czynna: nonakog gamma*;
1 fiolka zawiera:
250 JM** nonakogu gamma, rekombinowanego czynnika krzepnięcia krwi ludzkiej IX (rDNA), co odpowiada stężeniu 50 JM/ml po odtworzeniu leku za pomocą 5 ml roztwornika;
500 JM** nonakogu gamma, rekombinowanego czynnika krzepnięcia krwi ludzkiej IX (rDNA), co odpowiada stężeniu 100 JM/ml po odtworzeniu leku za pomocą 5 ml roztwornika;
1000 JM** nonakogu gamma, rekombinowanego czynnika krzepnięcia krwi ludzkiej IX (rDNA), co odpowiada stężeniu 200 JM/ml po odtworzeniu leku za pomocą 5 ml roztwornika;
2000 JM** nonakogu gamma, rekombinowanego czynnika krzepnięcia krwi ludzkiej IX (rDNA), co odpowiada stężeniu 400 JM/ml po odtworzeniu leku za pomocą 5 ml roztwornika;
3000 JM** nonakogu gamma, rekombinowanego czynnika krzepnięcia krwi ludzkiej IX (rDNA), co odpowiada stężeniu 600 JM/ml po odtworzeniu leku za pomocą 5 ml roztwornika.
substancje pomocnicze: L-histydyna, chlorek sodu, chlorek wapnia, manitol, sacharoza, polisorbat 80.
1 fiolka z roztwornikiem zawiera: wody do wstrzykiwań – 5 ml.
_________________________________________________________________________
* Nonakog gamma (rekombinowany czynnik krzepnięcia krwi IX (rDNA)) – to oczyszczony jednolancuchowy glikoprotein, zawierający 415 aminokwasów. Wytwarzany jest metodą rekombinowanego DNA w linii komórek jajnika chomika chińskiego.
** Aktywność (JM) określana jest metodą jednostopniowego analizy czynników krzepnięcia krwi zgodnie z Europejską Farmakopeą. Aktywność specyficzna leku RIXUBIS wynosi około 200–390 JM/mg białka.
Postać leku. Proszek i roztwornik do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizykochemiczne: proszek biały lub prawie biały; roztwornik – przezroczysty, bezbarwny roztwór.
Grupa farmakoterapeutyczna. Środki antyhemoragiczne. Czynnik krzepnięcia krwi IX.
Kod ATC B02B D04.
Właściwości farmakologiczne.
Farmakodynamika.
Mechanizm działania
Preparat Rixubis zawiera rekombinowany czynnik krzepnięcia krwi IX (nonakog gamma). Czynnik IX jest jednolancuchowym glikoproteidą o masie cząsteczkowej około 68 000 Daltonów. Jest witaminą K-zależnym czynnikiem krzepnięcia krwi, wytwarzanym w wątrobie. Czynnik IX jest aktywowany przez czynnik XIa we wewnętrznej drodze krzepnięcia oraz przez kompleks czynnika VII/tkankowego czynnika tkankowego w zewnętrznej drodze krzepnięcia. Aktywowany czynnik IX w połączeniu z aktywowanym czynnikiem VIII aktywuje czynnik X. Aktywowany czynnik X przekształca protrombinę w trombinę. Następnie trombina przekształca fibrynogen w fibrynę, w wyniku czego powstaje skrzeplina.
Farmakodynamika
Hemofilia B jest związana z płcią wrodzoną patologią układu krzepnięcia krwi, występującą na skutek obniżonych poziomów czynnika IX, prowadzącą do nasilonych krwawień do stawów, mięśni lub narządów wewnętrznych, spontanicznie lub w wyniku urazu przypadkowego lub chirurgicznego. Leczenie zastępcze pozwala na podniesienie poziomu czynnika IX we krwi i tym samym zapewnia tymczasową korekcję deficytu czynnika, zmniejszając skłonność do krwawień.
Skuteczność kliniczna i bezpieczeństwo
Profilaktyka i kontrola krwawień u pacjentów w wieku od 12 lat, którzy wcześniej poddawali się leczeniu
Skuteczność preparatu Rixubis oceniano w ramach niekontrolowanej części otwartego badania klinicznego fazy 1/3, w którym 73 pacjentów w wieku 12–59 lat, którzy wcześniej poddawali się leczeniu, otrzymywało preparat Rixubis w celu profilaktyki i/lub leczenia epizodów krwawień „na żądanie”. Wszyscy pacjenci mieli ciężką formę (poziom czynnika IX < 1%) lub umiarkowanie ciężką formę (poziom czynnika IX < 2%) hemofilii B. Spośród nich 59 osób otrzymywało preparat Rixubis w celu profilaktyki. Dane uzyskane u 56 pacjentów, którzy otrzymywali preparat Rixubis przez co najmniej 3 miesiące, zostały uwzględnione w zbiorze danych przeznaczonym do oceny skuteczności profilaktyki. Dodatkowo 14 pacjentów otrzymywało preparat Rixubis wyłącznie w celu leczenia epizodów krwawień. Pacjenci z kohorty leczenia „na żądanie” musieli mieć co najmniej 12 dokumentalnie potwierdzonych epizodów krwawień wymagających leczenia w ciągu 12 miesięcy poprzedzających włączenie do badania. Średnia długość leczenia pacjentów z kohorty leczenia „na żądanie” wynosiła 3,5 ± 1,00 miesiąca (mediana 3,4; zakres od 1,2 do 5,1 miesiąca), średnia całkowita częstość występowania krwawień w ciągu roku (ang. annualised bleeding rate (ABR)) wynosiła 33,9 ± 17,37 z medianą 27,0 i zakresem od 12,9 do 73,1.
Mediana ABR przy profilaktyce z zastosowaniem preparatu Rixubis dla wszystkich krwawień wynosiła 2,0, dla krwawień spontanicznych – 0,0 oraz dla krwawień do stawów – 0,0. U 24 pacjentów (42,9 %) nie stwierdzono krwawień.
Łącznie 249 epizodów krwawień zostało leczonych przy użyciu preparatu Rixubis, z czego 197 to krwawienia do stawów, a 52 – krwawienia pozastawowe (w tkankach miękkich, mięśniach, jamach ciała, przestrzeniach wewnątrzczaszkowych oraz inne). Spośród 249 epizodów krwawień 163 miało umiarkowaną ciężkość, 71 – niewielką, a 15 – ciężką. Leczenie prowadzono z uwzględnieniem indywidualnych cech każdego pacjenta, biorąc pod uwagę stopień ciężkości, przyczynę i lokalizację krwawienia. Większość (211 epizodów; 84,7 %) z 249 epizodów krwawień została wyleczona za pomocą 1–2 infuzji. Efektywność hemostatyczna w zatrzymaniu krwawienia oceniano jako doskonałą lub wysoką w 96 % wszystkich leczonych epizodów krwawień.
Profilaktyka i kontrola krwawień u pacjentów w wieku poniżej 12 lat, którzy wcześniej poddawali się leczeniu
Skuteczność preparatu Rixubis oceniano w ramach połączonego badania klinicznego fazy 2/3, w którym łącznie 23 pacjentów płci męskiej w wieku 1,8–11,8 roku (mediana wieku: 7,10 roku), którzy wcześniej poddawali się leczeniu, w tym 11 pacjentów w wieku poniżej 6 lat, otrzymywało preparat Rixubis w celu profilaktyki i kontroli epizodów krwawień. Wszyscy pacjenci mieli ciężką (poziom czynnika IX < 1 %) lub umiarkowanie ciężką (poziom czynnika IX < 2 %) formę hemofilii B. Wszyscy 23 pacjenci otrzymywali leczenie profilaktyczne preparatem Rixubis przez co najmniej 3 miesiące i zostali uwzględnieni w analizie oceny skuteczności profilaktyki.
Mediana ABR wynosiła 2,0, dla krwawień spontanicznych – 0,0 oraz dla krwawień do stawów – 0,0.
U dziewięciu pacjentów (39,1 %) nie stwierdzono krwawień.
Łącznie 26 epizodów krwawień zostało wyleczonych preparatem Rixubis, z czego 23 było spowodowane urazem, 2 – spontanicznie oraz 1 – o nieustalonej przyczynie. 19 krwawień występowało poza stawami (w tkankach miękkich, mięśniach, jamach ciała, przestrzeniach wewnątrzczaszkowych oraz inne), a 7 – to krwawienia do stawów, w tym jedno do stawu docelowego. Spośród wszystkich 26 epizodów krwawień 15 było niewielkiej ciężkości, 9 – umiarkowanej, a 2 – ciężkie. Leczenie prowadzono z uwzględnieniem indywidualnych cech każdego pacjenta, biorąc pod uwagę stopień ciężkości, przyczynę i miejsce krwawienia. Większość (23; 88,5 %) otrzymała leczenie w postaci 1–2 infuzji. Efektywność hemostatyczną w zatrzymaniu krwawienia oceniano jako doskonałą lub wysoką w 96,2 % wszystkich leczonych epizodów krwawień.
Leczenie perioperacyjne
Bezpieczeństwo i skuteczność stosowania preparatu Rixubis w warunkach leczenia perioperacyjnego oceniano w ramach wieloośrodkowego, prospektywnego, otwartego, niekontrolowanego badania fazy 3 z udziałem mężczyzn z ciężką i umiarkowaną hemofilią B, którzy wcześniej poddawali się leczeniu. Analiza skuteczności według protokołu obejmowała 37 zabiegów chirurgicznych, w tym duże i małe interwencje chirurgiczne, zabiegi stomatologiczne lub inne inwazyjne procedury chirurgiczne, przeprowadzone u 27 pacjentów w wieku od 17 do 57 lat. Dwadzieścia zabiegów było poważnych, w tym 13 zabiegów ortopedycznych i 3 stomatologiczne zabiegi chirurgiczne. 17 zabiegów, w tym 10 zabiegów wyłuskania zębów, uznano za niewielkie. Pacjenci, którym przeprowadzano duże zabiegi chirurgiczne, musieli przejść ocenę farmakokinetyki (FK). Wszyscy pacjenci otrzymywali dawkę dostosowaną do najnowszych danych dotyczących stopniowego przyrostu odbudowy aktywności czynnika IX. Zalecana dawka początkowa preparatu Rixubis miała zapewnić utrzymanie aktywności czynnika IX na poziomie 80–100 % podczas dużych zabiegów chirurgicznych i na poziomie 30–60 % podczas małych zabiegów. Preparat Rixubis podawano w formie infuzji bolusowej.
W całym okresie badania zapewniano utrzymanie hemostazy.
Farmakokinetyka
Pacjenci w wieku od 12 lat, którzy wcześniej poddawali się leczeniu
Zrandomizowane, ślepe, kontrolowane badanie krzyżowe farmakokinetyki preparatu Rixubis i preparatu porównawczego przeprowadzono u mężczyzn w stanie bez krwawienia (w wieku od 15 lat) w ramach centralnego połączonego badania klinicznego fazy 1/3. Pacjenci otrzymywali jeden lub drugi preparat w postaci pojedynczej infuzji dożylnej. Średnia (± odchylenie standardowe) i mediana dawki preparatu Rixubis w zbiorze danych analizy według protokołu (n = 25) wynosiły odpowiednio 74,69 ± 2,37 i 74,25 J/mkg z zakresem od 71,27 do 79,38 J/mkg. Parametry farmakokinetyczne obliczano na podstawie pomiarów aktywności czynnika IX w próbkach krwi pobranych w ciągu 72 godzin po każdej infuzji.
Ocenę farmakokinetyki preparatu Rixubis powtórzono w trakcie otwartego, niekontrolowanego badania z udziałem mężczyzn, którzy brali udział w pierwotnym badaniu krzyżowym FK i którzy otrzymywali profilaktykę preparatem Rixubis przez 26 ± 1 tydzień (średnio ± odchylenie standardowe) z całkowitym okresem stosowania preparatu Rixubis co najmniej 30 dni. Zakres dawek preparatu Rixubis stosowanych w trakcie powtórnego badania farmakokinetyki wynosił od 64,48 do 79,18 J/mkg (n = 23).
W tabeli 1 przedstawiono parametry farmakokinetyczne u wszystkich pacjentów, których dane można było ocenić (analiza według protokołu).
Tabela 1
| Parametr |
Rixubis Początkowe badanie krzyżowe (N = 25) |
Rixubis Ponowna ocena (N = 23) |
| AUC0-72 godz (MO • godz/dl)a Średnia ± odchylenie standardowe (OS) Mediana (zakres) |
1067,81 ± 238,42 1108,35 (696,07–1571,16) |
1156,15 ± 259,44 1170,26 (753,85–1626,81) |
| Wzrost odbudowy poziomu aktywności przy Cmax (MO/dl : MO/kg)b Średnia ± OS Mediana (zakres) |
0,87 ± 0,22 0,88 (0,53–1,35) |
0,95 ± 0,25 0,93 (0,52–1,38) |
| Okres półtrwania (godz) Średnia ± OS Mediana (zakres) |
26,70 ± 9,55 24,58 (15,83–52,34) |
25,36 ± 6,86 24,59 (16,24–42,20) |
| Cmax (MO/dl) Średnia ± OS Mediana (zakres) |
66,22 ± 15,80 68,10 (41,70–100,30) |
72,75 ± 19,73 72,40 (38,50–106,30) |
| Średni czas utrzymywania w organizmie (godz) Średnia ± OS Mediana (zakres) |
30,82 ± 7,26 28,93 (22,25–47,78) |
29,88 ± 4,16 29,04 (21,32–37,52) |
| Vssc (dl/kg) Średnia ± OS Mediana (zakres) |
2,02 ± 0,77 1,72 (1,10–3,94) |
1,79 ± 0,45 1,74 (1,12–2,72) |
| Klirens (dl/kg•godz) Średnia ± OS Mediana (zakres) |
0,0644 ± 0,0133 0,0622 (0,0426–0,0912) |
0,0602 ± 0,0146 0,0576 (0,0413–0,0945) |
a) Stężenie w osoczu – pole pod krzywą czasową w okresie 0–72 godziny po wlewie.
b) Obliczone jako wskaźnik (Cmax czynnika IX przed rozpoczęciem leczenia) podzielony przez dawkę w jednostkach MI/kg, gdzie Cmax – maksymalny poziom aktywności czynnika IX po wlewie.
c) Objętość rozłożenia w stanie ustalonym.
Wzrost odbudowy aktywności czynnika IX po 30 minutach od wlewu określano u wszystkich pacjentów włączonych do połączonego badania fazy 1/3 w dniu pierwszego leczenia, podczas wizyt u lekarza w 5., 13. i 26. tygodniu oraz w momencie zakończenia badania lub zaprzestania udziału w badaniu, jeśli nie pokrywa się on z wizytą w 26. tygodniu. Uzyskane dane wskazują, że wzrost odbudowy aktywności przez cały okres obserwacji jest stabilny (patrz tabela 2).
Tabela 2
| Wskaźnik |
1. dzień leczenia (N = 73) |
5. tydzień (N = 71) |
13. tydzień (N = 68) |
26. tydzień (N = 55) |
Dzień zakończenia/ przerwania badania b (N = 23) |
| Przyrost odbudowanej aktywności po 30 minutach od infuzji (j.m./dl : j.m./kg)a Średnia ± SD Mediana (zakres) |
0,79 ± 0,20 0,78 (0,26–1,35) |
0,83 ± 0,21 0,79 (0,46–1,48) |
0,85 ± 0,25 0,83 (0,14–1,47) |
0,89 ± 0,12 0,88 (0,52–1,29) |
0,87 ± 0,20 0,89 (0,52–1,32) |
a Obliczono jako wartość (C30 min czynnika IX przed rozpoczęciem leczenia) podzieloną przez dawkę w j.m./kg, gdzie C30 min to wartość pomiaru aktywności czynnika IX po 30 minutach od infuzji.
b Jeśli nie pokrywa się z czasem wizyty w 26. tygodniu.
Populacja pediatryczna (wcześniej leczona, poniżej 12. roku życia)
W ramach połączonego badania fazy 2/3 u 23 chłopców w wieku poniżej 12 lat, u których nie stwierdzono krwawienia, przeprowadzono wstępną ocenę farmakokinetyki leku Rixubis. Aby zmniejszyć dyskomfort związany z częstym pobieraniem próbek krwi u każdego pacjenta, pacjentów przydzielono losowo do dwóch grup z różnymi sekwencjami pobierania próbek krwi. Średnia dawka (± odchylenie standardowe) oraz mediana dawki leku Rixubis w pełnym zbiorze danych analitycznych (n = 23) wynosiły odpowiednio 75,50 ± 3,016 oraz 75,25 j.m./kg, w zakresie od 70,0 do 83,6 j.m./kg. Parametry farmakokinetyczne obliczono na podstawie pomiarów aktywności czynnika IX w próbkach krwi pobranych w ciągu 72 godzin od infuzji.
W Tabeli 3 przedstawiono parametry farmakokinetyczne dla wszystkich pacjentów (pełny zbiór danych analitycznych).
Tabela 3
| Parametr |
Do 6 lat (N = 11) |
Od 6 do 12 lat (N = 12) |
Wszyscy (N = 23) |
| AUCinf (MO • god/dl)a Średnia ± SD Mediana (zakres) |
723,7 ± 119,00 717,2 (488–947) |
886,0 ± 133,66 863,7 (730–1138) |
808,4 ± 149,14 802,9 (488–1138) |
| Okres półtrwania (godz.) Średnia ± SD Mediana (zakres) |
27,67 ± 2,66 27,28 (24,0–32,2) |
23,15 ± 1,58 22,65 (21,8–27,4) |
25,31 ± 3,13 24,48 (21,8–32,2) |
| Średni czas utrzymywania w organizmie (godz.) Średnia ± SD Mediana (zakres) |
30,62 ± 3,27 30,08 (26,2–36,2) |
25,31 ± 1,83 24,74 (23,7–30,3) |
27,85 ± 3,73 26,77 (23,7–36,2) |
| Vssb (dl/kg) Średnia ± SD Mediana (zakres) |
3,22 ± 0,52 3,16 (2,65–4,42) |
2,21 ± 0,32 2,185 (1,70–2,70) |
2,7 ± 0,67 2,69 (1,70–4,42) |
| Clearance (dl/kg • godz.) Średnia ± SD Mediana (zakres) |
0,1058 ± 0,01650 0,1050 (0,081–0,144) |
0,0874 ± 0,01213 0,0863 (0,069–0,108) |
0,0962 ± 0,01689 0,0935 (0,069–0,144) |
a Pole pod krzywą „stężenie w osoczu–czas” od 0 godzin do nieskończoności.
b Objętość rozkładu w stanie ustalonym.
Wartość przyrostu aktywności czynnika IX po 30 minutach od momentu infuzji była oceniana u wszystkich pacjentów włączonych do połączonego badania fazy 2/3 podczas oceny wstępnych parametrów farmakokinetycznych (w dniu pierwszego dnia leczenia), w dniu wizyt lekarskich w 5., 13. i 26. tygodniu oraz w momencie zakończenia badania lub wycofania się z badania, jeśli nie pokrywał się on z wizytą w 26. tygodniu. Uzyskane dane wskazują, że wartość przyrostu aktywności przez cały okres obserwacji była stabilna we wszystkich grupach wiekowych pediatrycznych (patrz tabele 4, 5, 6 poniżej).
Tabela 4
Wartość przyrostu aktywności po podaniu leku Rixubis po 30 minutach od momentu infuzji u dzieci z obu grup wiekowych
| Przyrost odbudowy aktywności po 30 minutach od infuzji |
FC (1 dzień leczenia) Wszyscy (N = 22) |
5 tydzień Wszyscy (N = 23) |
13 tydzień Wszyscy (N = 21) |
26 tydzień Wszyscy (N = 21) |
| (MO/dl: MO/kg)a Średnia ± SD Mediana (zakres) |
0,67 ± 0,16 0,69 (0,31–1,00) |
0,68 ± 0,12 0,66 (0,48–0,92) |
0,71 ± 0,13 0,66 (0,51–1,00) |
0,72 ± 0,15 0,734 (0,51–1,01) |
a Obliczono jako stosunek poziomu czynnika IX po 30 minutach (C30 min) przed rozpoczęciem leczenia do dawki w jednostkach MI/kg, gdzie C30 min to poziom aktywności czynnika IX zmierzony po 30 minutach od infuzji.
Tabela 5
Wzrost aktywności po zastosowaniu leku Rixubis po 30 minutach od infuzji u dzieci w wieku do 6 lat
| Przyrost odbudowy aktywności po 30 minutach po infuzji |
FVIII (1 dzień leczenia) Wszyscy (N = 10) |
5 tydzień, Wszyscy (N = 11) |
13 tydzień, Wszyscy (N = 10) |
26 tydzień, Wszyscy (N = 10) |
| (jedn./dl: jedn./kg) a Średnia ± SD Mediana (zakres) |
0,59 ± 0,13 0,59 (0,31–0,75) |
0,63 ± 0,10 0,6 (0,49–0,80) |
0,68 ± 0,12 0,66 (0,51–0,84) |
0,65 ± 0,13 0,61 (0,51–0,84) |
a Obliczono jako wskaźnik (C30 min czynnika IX przed rozpoczęciem leczenia) podzielony przez dawkę w jednostkach MI/kg, gdzie C30 min to wartość pomiaru aktywności czynnika IX po 30 minutach od infuzji.
Tabela 6
Wartości wzrostu aktywności czynnika IX po podaniu leku Rixubis po 30 minutach od infuzji u pacjentów w wieku od 6 do 12 lat
| Przyrost odbudowy aktywności po 30 min od infuzji |
FC (1 dzień leczenia) Wszyscy (N = 12) |
5. tydzień, Wszyscy (N = 12) |
13. tydzień, Wszyscy (N = 11) |
26. tydzień, Wszyscy (N = 11) |
| (MO/dl: MO/kg)a Średnia ± SD Mediana (zakres) |
0,73 ± 0,16 0,71 (0,51–1,00) |
0,73 ± 0,13 0,70 (0,48–0,92) |
0,73 ± 0,14 0,70 (0,54–1,00) |
0,8 ± 0,14 0,78 (0,56–1,01) |
a Obliczono jako stosunek stężenia czynnika IX po 30 minutach (C30 min czynnika IX przed rozpoczęciem leczenia) do dawki w jednostkach MI/kg, gdzie C30 min to wartość pomiaru aktywności czynnika IX po 30 minutach od infuzji.
Właściwości kliniczne.
Wskazania.
Do leczenia i zapobiegania krwawieniom u pacjentów z hemofilią B (wrodzony niedobór czynnika IX).
Lek Rixubis wskazany jest pacjentom z wszystkich grup wiekowych.
Przeciwwskazania.
Podwyższona wrażliwość na substancję czynną lub dowolny składnik pomocniczy wymieniony w sekcji „Skład”.
Znana reakcja alergiczna na białka chomika.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie zgłaszano interakcji leków opartych na ludzkim czynniku IX krzepnięcia krwi (rDNA) z innymi lekami.
Szczególne środki ostrożności.
Śledzenie
W celu poprawy śledzenia produktów biologicznych należy dokładnie odnotować nazwę i numer serii zastosowanego leku, np. w dzienniku osobistym.
Podwyższona wrażliwość
Zgłaszano reakcje nadwrażliwości o charakterze alergicznym podczas stosowania leku Rixubis. Preparat zawiera resztkową ilość białek chomika. W przypadku wystąpienia objawów nadwrażliwości należy zalecić pacjentom lub osobom opiekującym się nimi natychmiastowe zaprzestanie stosowania leku i skontaktowanie się z lekarzem. Pacjentów należy poinstruować o wczesnych objawach reakcji nadwrażliwości, w tym o pokrzywce, uogólnionej pokrzywce, uczuciu ściskania w klatce piersiowej, świstaniu podczas oddychania, hipotensji oraz anafilaksji.
Największe ryzyko występuje na wczesnych etapach stosowania skoncentrowanego czynnika IX u pacjentów wcześniej nieleczonych, szczególnie u pacjentów z wysokim ryzykiem mutacji genetycznych. W publikacjach zgłaszano związek między pojawieniem się inhibitora czynnika IX a reakcjami alergicznymi, szczególnie u pacjentów z wysokim ryzykiem mutacji genetycznych. Z tego powodu pacjentów z reakcjami alergicznymi należy kontrolować pod kątem obecności takiego inhibitora.
W przypadku wystąpienia wstrząsu należy podjąć standardowe działania medyczne w celu jego leczenia.
Inhibitory
Po ponownym leczeniu lekami opartymi na czynniku krzepnięcia krwi IX (rekombinowanym DNA) pacjentów należy badać pod kątem powstawania przeciwciał neutralizujących (inhibitorów), których stężenie należy oznaczać w jednostkach Bethesda (BU) przy użyciu odpowiedniej metody biologicznej.
W publikacjach zgłaszano związek między pojawieniem się inhibitora czynnika IX a reakcjami alergicznymi. Z tego powodu pacjentów z reakcjami alergicznymi należy kontrolować pod kątem obecności takiego inhibitora. Pacjenci, u których pojawiły się inhibitory czynnika IX, mają zwiększone ryzyko rozwoju anafilaksji w przypadku ponownego leczenia czynnikiem IX.
Z uwagi na ryzyko reakcji alergicznych podczas stosowania skoncentrowanego czynnika IX, wstępne podawanie czynnika IX powinno być, według uznania lekarza leczącego, przeprowadzane pod nadzorem medycznym, umożliwiającym zapewnienie odpowiedniej pomocy medycznej w przypadku wystąpienia reakcji alergicznych.
Zespół nerczycowy
Po stymulacji indukcji tolerancji immunologicznej u pacjentów z hemofilią typu B, u których występowały inhibitory czynnika IX, odnotowano przypadki rozwoju zespołu nerczycowego.
Zakrzepica
Ze względu na potencjalne ryzyko powikłań zakrzepowych należy wprowadzić kliniczne monitorowanie w celu wczesnego wykrywania objawów zakrzepicy i wyczerpującej koagulopatii poprzez odpowiednie badania biologiczne podczas stosowania tego leku u pacjentów z chorobą wątroby, po operacjach, u noworodków lub u pacjentów z ryzykiem rozwoju zjawisk zakrzepowych lub zespołem rozsianej wewnątrznaczyniowej krzepnięcia (DIC). W każdym z tych przypadków należy rozważyć korzyści wynikające z leczenia lekiem Rixubis w porównaniu z ryzykiem wystąpienia takich powikłań.
Choroby układu sercowo-naczyniowego
U pacjentów z istniejącymi chorobami układu sercowo-naczyniowego terapia zastępcza z zastosowaniem czynnika IX może zwiększyć ryzyko rozwoju patologii sercowo-naczyniowej.
Powikłania związane z zastosowaniem kaniuli
W razie konieczności zastosowania urządzenia dożylnego centralnego (CVC) należy wziąć pod uwagę możliwość wystąpienia powikłań związanych z CVC, w tym lokalnych infekcji, bakteriemii i zakrzepicy w miejscu wprowadzenia kaniuli.
Uwagi dotyczące obecności substancji pomocniczych
Po odtworzeniu, ten lek zawiera mniej niż 1 mmol (23 mg) sodu w jednym fiolce, czyli praktycznie nie zawiera sodu. W zależności od masy ciała pacjenta i zalecanego dawkowania pacjenci mogą otrzymać więcej niż jedną fiolkę leku Rixubis, co należy uwzględnić w przypadku przestrzegania diety z kontrolowaną zawartością sodu.
Pacjenci w wieku podeszłym
Pacjenci w wieku 65 lat i starsi nie byli rekrutowani do badań klinicznych leku Rixubis. Nie wiadomo, czy ich reakcja na leczenie różni się od reakcji młodszych pacjentów. Tak jak u wszystkich innych pacjentów, dobór dawki u pacjentów w wieku podeszłym należy przeprowadzać indywidualnie.
Dzieci
Wymienione powyżej ostrzeżenia i środki ostrożności dotyczą leczenia zarówno dorosłych, jak i dzieci.
Stosowanie w okresie ciąży lub karmienia piersią.
Ciąża
Dane dotyczące stosowania czynnika IX u kobiet w okresie ciąży są nieobecne lub ograniczone. Nie przeprowadzono badań wpływu czynnika IX na funkcję rozrodczą u zwierząt.
Czynnik IX należy stosować w okresie ciąży i karmienia piersią tylko wtedy, gdy wskazanie do jego stosowania jest wyraźne.
Karmienie piersią
Brak danych dotyczących wydzielania czynnika IX lub jego metabolitów z mlekiem matki.
Plodność
Brak informacji dotyczących wpływu czynnika IX na płodność.
Wpływ na zdolność prowadzenia pojazdów i obsługiwania maszyn.
Lek Rixubis nie wpływa na zdolność prowadzenia pojazdów lub obsługiwanie maszyn.
Sposób stosowania i dawki
Leczenie powinno być prowadzone pod nadzorem lekarza z doświadczeniem w leczeniu hemofilii.
Monitorowanie w trakcie leczenia
W trakcie leczenia zaleca się odpowiednie oznaczanie poziomów czynnika IX jako wytycznych do ustalania dawki i częstotliwości powtórzonych infuzji. Odpowiedź na czynnik IX może różnić się u poszczególnych pacjentów, z różnym okresem połowicznego rozpadu i stopniem odbudowy. Pacjentom o niskiej lub nadmiernej masie ciała może być konieczna korekta dawki obliczanej na podstawie masy ciała. W przypadku poważnych zabiegów chirurgicznych konieczne jest dokładne monitorowanie w trakcie terapii zastępczej poprzez analizę czynników krzepnięcia krwi (aktywność czynnika IX w osoczu).
W celu osiągnięcia pożądanych poziomów aktywności czynnika IX w osoczu krwi zaleca się staranne monitorowanie przy użyciu odpowiedniej metody oznaczania aktywności czynnika IX oraz, w razie potrzeby, odpowiednią korektę dawki i częstotliwości powtórzonych infuzji. W przypadku stosowania jednostopniowej analizy in vitro czynników krzepnięcia krwi według czasu tromboplastynowego częściowego (APTT) do oznaczania aktywności czynnika IX w próbkach krwi pacjentów, wyniki dotyczące aktywności czynnika IX mogą znacząco zależeć zarówno od rodzaju użytego odczynnika APTT, jak i od zastosowanego standardu porównawczego. Jest to szczególnie istotne przy zmianie laboratorium i/lub odczynników stosowanych do analizy.
Dawki
Wielkość dawki i długość trwania terapii zastępczej zależą od stopnia niedoboru czynnika IX, lokalizacji i nasilenia krwawienia, a także od stanu klinicznego, wieku i parametrów farmakokinetycznych czynnika IX u pacjenta, takich jak wzrost aktywności czynnika IX i okres połowicznego rozpadu.
Zastosowana ilość jednostek czynnika IX wyrażana jest w jednostkach międzynarodowych (j.m.) zgodnie z obowiązującym standardem WHO dla leków opartych na czynniku IX. Aktywność czynnika IX w osoczu krwi wyrażana jest w procentach (w odniesieniu do normalnej wartości dla osocza ludzkiego) lub w jednostkach międzynarodowych (zgodnie z międzynarodowym standardem dla czynnika IX w osoczu krwi).
Jedna jednostka międzynarodowa (j.m.) aktywności czynnika IX odpowiada aktywności czynnika IX w 1 ml normalnego osocza ludzkiego.
Dorośli
Leczenie „na żądanie”
Obliczenie wymaganej dawki czynnika IX dla pacjentów w wieku od 12 lat opiera się na empirycznych ustalenich, że podanie jednej jednostki międzynarodowej (j.m.) czynnika IX na 1 kg masy ciała prowadzi do wzrostu aktywności czynnika IX w osoczu o 0,9 j.m./dl (zakres od 0,5 do 1,4 j.m./dl) lub o 0,9 % aktywności normalnej.
Wymaganą dawkę określa się według poniższego wzoru.
| Potrzebna ilość jednostek |
= |
masa ciała (kg) |
× |
pożądane zwiększenie poziomu czynnika IX (% lub MI/dl) |
× |
odwrotność wartości zarejestrowanego odnowienia aktywności czynnika IX (dl/kg) |
W celu osiągnięcia stopniowego wzrostu poziomu aktywności o 0,9 MI/dl na 1 MI/kg dawkę oblicza się według następującego wzoru:
| Potrzebna ilość jednostek |
= |
masa ciała (kg) |
× |
pożądane zwiększenie poziomu czynnika IX (% lub JU/dl) |
× |
1,1 dl/kg |
Dawkę i częstotliwość planowanego stosowania należy zawsze dostosować tak, aby zapewnić skuteczność kliniczną u każdego poszczególnego pacjenta.
W przypadku wystąpienia krwawień wymienionych poniżej, poziom aktywności czynnika IX nie powinien być niższy niż wskazany poziom aktywności we krwi (w procentach normy lub MI/dl) przez odpowiedni okres czasu. Tabelę 7 można wykorzystać do ustalenia dawki w przypadku epizodów krwawień oraz podczas zabiegów chirurgicznych.
Tabela 7
| Stopień ciężkości krwawienia/ typ zabiegu chirurgicznego |
Wymagany poziom czynnika IX, % lub JE/dl |
Częstotliwość stosowania (godziny)/ czas trwania leczenia (dni) |
| Krwawienie Wczesny hemartroza, krwawienie do mięśni lub jamy ustnej |
20–40 |
Powtarzaj co 24 godziny. Przynajmniej 1 dzień, aż do ustania krwawienia, o czym świadczy brak bólu, lub do gojenia się rany. |
| Bardziej nasilona hemartroza, krwawienie do mięśni lub guz krwawiowy |
30–60 |
Powtarzaj infuzję co 24 godziny przez 3–4 dni lub dłużej, aż do zniknięcia bólu i ustąpienia wyraźnych zaburzeń funkcjonalnych. |
| Krwawienia zagrażające życiu |
60–100 |
Powtarzaj infuzję co 8–24 godziny, aż do zniknięcia zagrożenia. |
| Zabiegi chirurgiczne Małe zabiegi chirurgiczne, w tym usuwanie zębów |
30–60 |
Co 24 godziny, przynajmniej 1 dzień, aż do gojenia się rany. |
| Duże zabiegi chirurgiczne |
80–100 (przed zabiegiem i po nim) |
Powtarzaj infuzję co 8–24 godziny, aż do odpowiedniego gojenia się rany, a następnie kontynuuj leczenie przynajmniej przez kolejne 7 dni, utrzymując aktywność czynnika IX na poziomie 30–60% (JE/dl). |
Ścisła kontrola przebiegu leczenia zastępczego jest szczególnie ważna w przypadku poważnych zabiegów chirurgicznych lub krwawienia stanowiącego zagrożenie dla życia.
Profilaktyka
W celu długoterminowej profilaktyki krwawień u pacjentów w wieku od 12 lat z ciężką formą hemofilii B, stosuje się zazwyczaj dawki od 40 do 60 MI czynnika IX na 1 kg masy ciała co 3–4 dni. W niektórych przypadkach, w zależności od farmakokinetyki, wieku, fenotypu krwawienia oraz stopnia aktywności fizycznej pacjenta, może być konieczne skrócenie odstępu między dawkami lub zwiększenie dawki leku.
Ciągła infuzja
Nie podawać leku Rixubis w formie ciągłej infuzji.
Sposób podania
Podanie dożylnie.
W przypadku podawania leku przez samego pacjenta lub osobę opiekującą się pacjentem, należy odpowiednio wykonać szkolenie w zakresie wykonywania tej procedury.
Lek Rixubis należy podawać z taką szybkością, która zapewnia komfort dla pacjenta, maksymalnie do 10 ml/min.
Po odtworzeniu lek jest klarownym, bezbarwnym roztworem, bez zanieczyszczeń mechanicznych, o pH 6,8–7,2. Osmolalność wynosi ponad 240 mOsmol/kg.
Do podania tego leku należy używać wyłącznie plastikowych strzykawek z końcówką Luer.
Lek Rixubis podaje się dożylnie po odtworzeniu proszku w rozcieńczycielu dołączonym do opakowania.
- Do przygotowania roztworu leku należy używać wyłącznie rozcieńczyciela oraz urządzenia do odtworzenia leku (BAXJECT II) zawartych w opakowaniu.
- Do podania leku należy używać strzykawki z końcówką Luer.
- Nie należy używać urządzenia BAXJECT II, jeśli jest uszkodzone, jeśli uszkodzony jest jego sterylny system ochrony lub opakowanie, lub jeśli występują jakiekolwiek oznaki pogorszenia stanu.
Odtworzenie
Należy stosować metody aseptyczne.
- Jeśli lek był przechowywany w lodówce, wyjmij fiolki z proszkiem Rixubis oraz z rozcieńczycielem i pozostaw je do ogrzania do temperatury pokojowej (od 15 °C do 30 °C).
- Dokładnie umyj ręce ciepłą wodą z mydłem.
- Zdejmij pokrywki z fiolek z proszkiem i rozcieńczycielem.
- Przetrzyj septa tamponami z alkoholem. Umieść fiolki na równej, czystej powierzchni.
- Otwórz opakowanie z urządzeniem BAXJECT II, zdejmując papierową folię, nie dotykając wewnętrznej powierzchni urządzenia (rysunek a). Nie wyjmuj urządzenia z opakowania.
- Odwróć opakowanie i nałóż przezroczysty plastikowy sztyft na septum fiolki z rozcieńczycielem. Trzymając opakowanie za krawędzie, zdejmij opakowanie z urządzenia BAXJECT II (rysunek b). Nie zdejmuj niebieskiego kapturka z urządzenia BAXJECT II.
- Gdy urządzenie BAXJECT II jest już podłączone do fiolki z rozcieńczycielem, odwróć układ tak, aby fiolka z rozcieńczycielem znalazła się nad urządzeniem. Nałóż biały plastikowy sztyft na septum fiolki z lekiem Rixubis. Rozcieńczyciel zostanie automatycznie przesycany pod działaniem próżni do fiolki zawierającej lek Rixubis (rysunek c).
- Delikatnie wymieszaj, aż do całkowitego rozpuszczenia proszku. Lek rozpuszcza się szybko (w ciągu 2 minut). Upewnij się, że Rixubis został całkowicie rozpuszczony, ponieważ w przeciwnym przypadku nie cała odtworzona dawka leku przejdzie przez filtr urządzenia. Przed podaniem odtworzonego leku należy dokonać wizualnej kontroli w celu wykrycia cząstek stałych i zmian zabarwienia. Roztwór powinien być klarowny lub lekko opalescencyjny. Nie należy stosować roztworów, które są zmętniałe lub zawierają osad.
| Rysunek a |
Rysunek b |
Rysunek c |
|
|
||
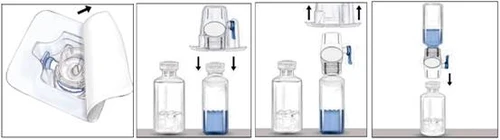
Nie chłodzić leku po odtworzeniu. Należy go użyć natychmiast po przygotowaniu.
Zastosowanie
Stosować technikę aseptyczną.
- Zdejmij niebieską nakładkę z urządzenia BAXJECT II. Zadbaj o to, aby nie wciągnąć powietrza do strzykawki. Dołącz strzykawkę do urządzenia BAXJECT II (rysunek d).
- Odwróć układ (fiolka z odtworzonym roztworem powinna znajdować się u góry). Napełnij odtworzonym roztworem strzykawkę, powoli wyciągając tłok (rysunek e).
- Odłącz strzykawkę.
- Dołącz igłę motylkową do strzykawki. Wprowadź roztwór do żyły. Roztwór należy podawać powoli, z szybkością komfortową dla pacjenta, nie szybciej niż 10 ml na minutę.
| Rysunek d |
Rysunek e |
|
|
|
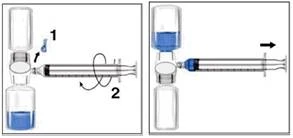
Zawsze podczas stosowania leku Rixubis należy zapisywać jego nazwę i numer serii (np. w dzienniku osobistym), aby zachować informacje o stosowanych lekach.
Środki, które nie zostały wykorzystane, oraz wszelkie odpady należy utylizować zgodnie z lokalnymi przepisami.
Stabilność chemiczna i fizyczna rozcieńczonego, gotowego do użycia leku wynosi 3 godziny w temperaturze nie wyższej niż 30 °C. Z mikrobiologicznego punktu widzenia, mimo że sposób rozcieńczania wyklucza zakażenie mikrobiologiczne, lek należy stosować natychmiast. Jeżeli lek nie zostanie zastosowany natychmiast, okres i warunki przechowywania są odpowiedzialnością użytkownika. Nie chłodzić.
Dzieci
Pacjenci w wieku od 12 do 17 lat
Dawkowanie jest takie samo jak u dorosłych i dzieci w wieku od 12 do 17 lat.
Pacjenci w wieku poniżej 12 lat
Leczenie „na żądanie”
Dawkę czynnika IX dla pacjentów w wieku poniżej 12 lat oblicza się na podstawie doświadczeń empirycznych, zgodnie z którymi podanie jednej jednostki międzynarodowej (j.m.) czynnika IX na 1 kg masy ciała prowadzi do wzrostu aktywności czynnika IX we krwi do poziomu 0,7 j.m./dl (zakres od 0,31 do 1,0 j.m./dl) lub do 0,7 % aktywności normalnej.
Wymaganą dawkę oblicza się według poniższego wzoru.
| Potrzebna ilość jednostek |
= |
masa ciała (kg) |
× |
pożądane zwiększenie poziomu czynnika IX (% lub MI/dl) |
× |
odwrotność wartości zarejestrowanego odsetka odbudowy aktywności (dl/kg) |
W celu stopniowego wzrostu poziomu aktywności o 0,7 MI/dl na 1 MI/kg dawkowanie oblicza się według następującego wzoru:
| Potrzebna ilość jednostek |
= |
masa ciała (kg) |
× |
pożądany wzrost poziomu czynnika IX (% lub MI/dl) |
× |
1,4 dl/kg |
Do dawkowania w przypadkach krwawień oraz podczas zabiegów chirurgicznych można wykorzystać tę samą tabelę, co dla dorosłych (patrz powyżej tabela 7).
Profilaktyka
Zakres zalecanych dawek stosowanych u dzieci w wieku do 12 lat wynosi 40–80 JМ/ kg co 3–4 dni. W niektórych przypadkach, w zależności od farmakokinetyki, wieku, fenotypu krwawienia oraz poziomu aktywności fizycznej pacjenta, może być konieczne skrócenie odstępu między dawkami lub zwiększenie dawki leku.
Nadmierna dawka.
Nie opisano skutków działania dawek wyższych niż zalecane preparatu Rixubis.
Efekty uboczne
Obserwowano rzadkie przypadki nadwrażliwości lub reakcji alergicznych (mogące obejmować obrzęk naczynioruchowy, uczucie pieczenia i swędzenia w miejscu infuzji, dreszcze, zarzut krwi do twarzy, uogólnione pokrzywki, ból głowy, wysypkę, hipotensję, letargię, nudności, stan niepokoju, tachykardię, uczucie ściskania w klatce piersiowej, dzwonienie w uszach, wymioty, świsty podczas oddychania), które czasem mogły postępować do ciężkiej formy anafilaksji (w tym wstrząs), co występowało w ścisłym związku czasowym z pojawieniem się inhibitorów czynnika IX.
Po stymulacji indukcji tolerancji immunologicznej u pacjentów z hemofilią typu B i z wcześniejszymi reakcjami alergicznymi, u których występowały inhibitory czynnika IX, odnotowano przypadki zespołu nerczycowego.
Bardzo rzadko występują przypadki powstawania przeciwciał przeciwko białkom chomika, towarzyszące reakcjom nadwrażliwości.
U pacjentów z hemofilią typu B mogą powstawać przeciwciała neutralizujące (inhibitory) przeciwko czynnikowi IX, co objawia się niewystarczającą skutecznością leczenia. W takich przypadkach zaleca się skonsultowanie się ze specjalistycznym ośrodkiem leczenia hemofilii.
Stosowanie leków opartych na czynniku IX wiąże się z potencjalnym ryzykiem wystąpienia epizodów zakrzepowo-zatorowych, przy czym ryzyko to jest większe przy stosowaniu preparatów o niskim stopniu oczyszczenia. Stosowanie preparatów czynnika IX o niskim stopniu oczyszczenia wiązało się z przypadkami zawału mięśnia sercowego, rozsianej wewnątrzwatrobnej krzepnięcia krwi (DIC), zakrzepicy żył głębokich i zatorowości płucnej. Stosowanie preparatów czynnika IX o wysokim stopniu oczyszczenia rzadko wiąże się z wystąpieniem takich efektów ubocznych.
Lista efektów ubocznych w formie tabeli
W badaniach klinicznych dotyczących stosowania leku Rixubis wzięło udział 99 osób, z których co najmniej jedna osoba otrzymała lek, co wiązało się z wystąpieniem łącznie 5 efektów ubocznych. Poniższa tabela zawiera informacje o efektach ubocznych według klas systemów i narządów (KSN) według klasyfikacji MedDRA (przy użyciu terminów preferowanego użytkowania).
Częstotliwość występowania efektów ubocznych sklasyfikowano według następującego kryterium: bardzo często (> 1/10), często (od > 1/100 do < 1/10), rzadko (od > 1/1000 do < 1/100), rzadko (od > 1/10000 do < 1/1000), bardzo rzadko (< 1/10000), częstotliwość nieznana (na podstawie dostępnych danych niemożliwe jest oszacowanie częstotliwości).
W ramach każdej kategorii częstotliwości efekty uboczne przedstawiono w kolejności malejącej ciężkości.
Tabela 8
| Pozwiązane reakcje niepożądane zarejestrowane w badaniach klinicznych i zgłoszeniach spontanicznych |
||
| Klasa układów i narządów według klasyfikacji MedDRA |
Reakcje niepożądane |
Częstość występowania na jednego pacjenta |
| Zaburzenia układu odpornościowego |
Zwiększona wrażliwość a) |
Nieznana |
| Zaburzenia układu nerwowego |
Zaburzenia smaku |
Często |
| Zaburzenia układu mięśniowo-szkieletowego i tkanki łącznej |
Ból kończyn |
Często |
a) PR (reakcja niepożądana) wyjaśniona jest poniżej w sekcji.
Opis poszczególnych reakcji niepożądanych
Podwyższona wrażliwość
Reakcje alergiczne występowały w postaci duszności, świądu, uogólnionej pokrzywki i wysypek.
Dzieci
Oczekuje się, że częstość, rodzaj i ciężkość reakcji niepożądanych u dzieci będą takie same jak u dorosłych. Dane dotyczące nieleczonych pacjentów są jednak niedostępne, ponieważ w badaniach klinicznych uczestniczyli wyłącznie pacjenci wcześniej leczeni. W związku z tym nie przeprowadzono badań immunogenności w kontekście powstawania inhibitorów u pacjentów z tej grupy ryzyka.
Zgłaszanie podejrzewanych reakcji niepożądanych
Zgłaszanie podejrzewanych reakcji niepożądanych po rejestracji leku ma istotne znaczenie. Umożliwia to monitorowanie stosunku korzyści do ryzyka w przypadku stosowania tego leku. Personel medyczny i farmaceutyczny, a także pacjenci lub ich ustawowi przedstawiciele powinni zgłaszać wszystkie przypadki podejrzewanych reakcji niepożądanych oraz braku skuteczności leku za pośrednictwem Zautomatyzowanego Systemu Informacyjnego do Nadzoru Farmakologicznego pod adresem: https://aisf.dec.gov.ua.
Okres ważności.
3 lata.
Warunki przechowywania.
Przechowywać w temperaturze nie wyższej niż 30 °C. Nie zamrażać.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Ze względu na brak badań dotyczących zgodności, tego leku nie wolno mieszać z innymi lekami.
Podczas stosowania tego leku można używać wyłącznie strzykawek plastikowych z końcówką typu Luer. Ze względu na adsorpcję czynnika IX krzepnięcia krwi człowieka na wewnętrznych powierzchniach niektórych urządzeń do infuzji dawkowanie leku może być nieprawidłowe.
Opakowanie.
1 fiolka z proszkiem (250 MI, 500 MI, 1000 MI, 1500 MI, 2000 MI lub 3000 MI) w zestawie z 1 fiolką rozpuszczalnika (5 ml wody do wstrzykiwań) oraz 1 urządzeniem do rozcieńczania BAKSJET II w opakowaniu kartonowym.
Kategoria wydawania.
Na receptę.
Producent.
Baxalta Belgia Wytwórstwo SA / Baxalta Belgium Manufacturing SA.
Miejsce produkcji oraz adres siedziby działalności.
Boulevard Rene Branquart 80, Lessines, 7860, Belgia / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.