Refaktor AF
UkrainaSpis treści
INSTRUKCJA do stosowania leczniczego leku Refaktor AF (ReFacto® AF)
Skład:
substancja czynna: moroctocog alfa (rekombinowany czynnik krzepnięcia krwi VIII);
1 fiolka zawiera 250 JМ lub 500 JМ lub 1000 JМ lub 2000 JМ moroctocogu alfa;
1 szpryceta wstępnie napełniona zawiera 3000 JМ moroctocogu alfa;
substancje pomocnicze: sacharoza, chlorek wapnia dwuwodny, L-histydyna, polisorbat 80, chlorek sodu.
Roztwórnik: chlorek sodu, woda do wstrzykiwań.
Postać leku.
Liofilizat do sporządzenia roztworu do wstrzykiwań.
Główne właściwości fizyko-chemiczne:
postać opakowania – fiolka:
liofilizat białego koloru, praktycznie wolny od wyraźnie widocznych zanieczyszczeń, wilgoci oraz wad uszczelnienia fiolki;
roztwórnik: bezbarwny, klarowny roztwór, praktycznie wolny od widocznych wtrąceń;
postać opakowania – szpryceta wstępnie napełniona:
w górnej komorze – liofilizat białego koloru, praktycznie wolny od wyraźnie widocznych zanieczyszczeń, wilgoci oraz wad uszczelnienia pojemnika;
w dolnej komorze – roztwórnik: bezbarwny, klarowny roztwór, praktycznie wolny od widocznych wtrąceń.
Grupa farmakoterapeutyczna.
Środki przeciwkrwotoczne. Czynnik krzepnięcia krwi VIII. Kod ATC B02B D02.
Właściwości farmakodynamiczne.
Farmakodynamika.
Refaktor AF zawiera rekombinowany czynnik krzepnięcia krwi VIII z usuniętym domeną B (moroctocog alfa). Aktywność leku w jednostkach międzynarodowych (JMi) określa się metodą analizy chromatogenicznej zgodnie z Europejską Farmakopeą. Aktywność specyficzna leku Refaktor AF wynosi 7 600–13 800 JMi/mg białka.
Moroctocog alfa to glikoproteina o przybliżonej masie cząsteczkowej 170 000 Da, składająca się z 1438 aminokwasów, której sekwencja odpowiada formie czynnika VIII o masie 90 + 80 kDa (tj. z usuniętą domeną B), a modyfikacje potranslacyjne są podobne do tych występujących w cząsteczce pochodzącej z osocza krwi.
Moroctocog alfa wytwarza się metodą rekombinowanego DNA przy użyciu komórek jajnika chomika chińskiego. Proces produkcji Refaktor AF został zmodyfikowany w taki sposób, aby wykluczyć możliwość zanieczyszczenia białkami pochodzenia ludzkiego lub zwierzęcego w trakcie hodowli komórkowej, oczyszczania lub produkcji produktu gotowego; jednocześnie nazwę leku zmieniono z Refaktor na Refaktor AF.
Właściwości funkcjonalne Refaktor AF są podobne do właściwości endogennego czynnika VIII. Aktywność czynnika VIII jest znacznie obniżona u pacjentów z hemofilią A, dlatego wymagają oni terapii zastępczej.
Po podaniu dożylnym czynnik VIII wiąże się z czynnikiem von Willebranda obecnym w krwiobiegu.
Aktywowany czynnik VIII działa jako kofaktor dla aktywowanego czynnika IX, przyspieszając przekształcanie czynnika X w aktywowany czynnik X. Aktywowany czynnik X przekształca protrombinę w trombinę. Trombina z kolei przekształca fibrynogen w fibrynę, w wyniku czego powstaje skrzep. Hemofilia A to warunkowana genetycznie, sprzężona z płcią choroba krwawicza spowodowana obniżonym poziomem czynnika VIII:C, prowadząca do obfitych krwawień do stawów, mięśni lub narządów wewnętrznych. Krwawienia mogą występować spontanicznie lub jako wynik urazów przypadkowych lub operacyjnych. W wyniku zastosowania terapii zastępczej poziom czynnika VIII w osoczu krwi wzrasta, co pozwala tymczasowo skorygować niedobór tego czynnika i zmniejszyć skłonność do krwawień.
Skuteczność kliniczna
Dane przedstawione w tabeli 1 pochodzą z badań stosowania leku Refaktor AF u pacjentów wcześniej nieleczonych (PNL) oraz u pacjentów wcześniej leczonych (PL), w wieku < 12 lat.
Tabela 1
Dawkowanie i skuteczność stosowania u dzieci
| Wskaźnik |
PLP |
PLP |
PNP |
| Dawkowanie wg masy ciała (JE/kg) do profilaktycznej infuzjia |
N = 14 (28, 51) |
N = 13 (21, 49) |
N = 22 (17, 161) |
| Ogólny wskaźnik częstości krwawień (OTR) u wszystkich podmiotów, |
-- |
-- |
N = 23 (0,0; 39,5) |
| Ogólny wskaźnik częstości krwawień u podmiotów stosujących reżim „na żądanie” na początku badaniac, |
N = 5 (1,6; 50,6) |
N = 9 (0,0; 46,6) |
-- |
| Ogólny wskaźnik częstości krwawień u podmiotów stosujących reżim „profilaktyczny” na początku badaniac, |
N = 13 (0,0; 11,2) |
N = 9 (0,0; 13,0) |
-- |
| Dawkowanie wg masy ciała (JE/kg) w epizodzie krwawienia do leczenia krwawienia, mediana (min., maks.) |
N = 13 (28; 86) |
N = 14 (17; 229) |
N = 21 (11; 221) |
| % krwawień, które zostały pomyślnie wyleczone za pomocą ≤ 2 infuzji |
98,7 % |
98,8 % |
96,7 % |
a Dawkowanie i częstotliwość stosowania leku Refaktor AF w badaniu określano na podstawie standardów terapeutycznych stosowanych lokalnie.
b Podmioty w badaniu PNP nie były zobowiązane do stosowania regularnej, ciągłej terapii profilaktycznej; jednakże, z wyjątkiem jednego podmiotu (któremu podawano wyłącznie leczenie „na żądanie”), większość podmiotów otrzymywała regularne infuzje profilaktyczne. Kilka podmiotów rozpoczęło udział w badaniu z infuzjami „na żądanie”, a następnie przeszło na leczenie profilaktyczne, niektóre podmioty otrzymywały jedynie okazjonalne infuzje profilaktyczne.
c Podmioty w badaniu PLP na początku badania wskazywały tryb terapii czynnikiem VIII („profilaktyka” lub „na żądanie”) i nie były zobowiązane do przestrzegania tego trybu jako warunku uczestnictwa w badaniu. Dawkowanie i częstotliwość stosowania leku Refaktor AF w badaniu określano przez lekarza na podstawie lokalnych standardów terapii.
Skróty: ŚRR – średni roczny poziom krwawień.
Należy zaznaczyć, że nie porównuje się wskaźnika ŚRR przy stosowaniu różnych koncentratów czynników krzepnięcia krwi oraz w różnych badaniach klinicznych.
Indukcja tolerancji immunologicznej.
Zebrano dane dotyczące indukcji tolerancji immunologicznej u pacjentów z hemofilią A, u których pojawiły się inhibitory czynnika VIII. W ramach podstawowego badania klinicznego Refaktora z udziałem 25 pacjentów, którzy wcześniej nie otrzymywali leczenia, zebrano dane dotyczące indukcji tolerancji immunologicznej (15 z wysokim mianem, 10 z niskim mianem). Spośród 25 tych pacjentów u 20 pacjentów miano obniżenie miana inhibitorów do poziomu < 0,6 JB (jednostek Bethesda), z czego u 11 spośród 15 początkowo obserwowano wysokie miana (≥ 5 JB/ml), a u 9 spośród 10 – niskie. Analogiczne obniżenie miana zaobserwowano u 5 spośród 6 pacjentów, u których stwierdzono niskie miana inhibitorów, ale nie podawano im leczenia polegającego na indukcji tolerancji immunologicznej. Obserwacji odległych skutków nie prowadzono.
Farmakokinetyka.
Właściwości farmakokinetyczne Refaktora, określone w badaniu krzyżowym Refaktora i koncentratu czynnika VIII pochodzącego z osocza krwi, z udziałem 18 pacjentów, którzy wcześniej otrzymywali leczenie (wcześniej leczeni pacjenci, PLP), przedstawiono w tabeli 2. Badanie ilościowe prowadzono z zastosowaniem podłoża chromogenowego (patrz sekcja „Sposób stosowania i dawki”).
Tabela 2
| Ocena parametrów farmakokinetycznych Refaktor AF u chorych na hemofilię typu A |
|||
| Parametr |
Średnia |
SD |
Mediana |
| AUCt (IU·godz/ml) |
19,9 |
4,9 |
19,9 |
| t½ (godz) |
14,8 |
5,6 |
12,7 |
| CL (ml/godz·kg) |
2,4 |
0,75 |
2,3 |
| średni czas utrzymywania (godz) |
20,2 |
7,4 |
18,0 |
| odzysk (IU/dl wzrost czynnika VIII:C na IU/kg zastosowanego czynnika VIII) |
2,4 |
0,38 |
2,5 |
Skróty: AUCt – pole pod krzywą „stężenie w osoczu – czas” od zera do ostatniego zmierzonego stężenia; t½ – okres półtrwania; CL – klirens; SD – odchylenie standardowe.
W badaniu, w którym porównywano aktywność Refaktor AF, Refaktor i czynnika VIII w osoczu krwi pacjentów metodą analizy chromogennej, stwierdzono bioekwiwalentność Refaktor AF i Refaktor. Stosunek średnich geometrycznych wartości parametrów farmakokinetycznych wyznaczonych metodą najmniejszych kwadratów dla Refaktor AF i Refaktor wynosił odpowiednio 100,6%, 99,5% i 98,1% dla odzysku, AUCt i AUC∞ (pole pod krzywą „stężenie w osoczu – czas” od zera do nieskończoności). Odpowiednie 90% przedziały ufności dla stosunku średnich geometrycznych wartości parametrów farmakokinetycznych Refaktor AF i Refaktor mieściły się w granicach okna bioekwiwalentności (od 80 do 125%), co świadczy o bioekwiwalentności Refaktor AF i Refaktor.
W krzyżowym badaniu farmakokinetycznym parametry farmakokinetyczne Refaktor AF określano u 25 pacjentów (≥ 12 lat), którzy wcześniej otrzymywali leczenie, na początku badania oraz po sześciu miesiącach wielokrotnego stosowania Refaktor AF. Stosunek średnich geometrycznych wartości parametrów farmakokinetycznych wyznaczonych metodą najmniejszych kwadratów po 6 miesiącach stosowania w porównaniu z wartościami wyjściowymi wynosił odpowiednio 107%, 100% i 104% dla odzysku, AUCt i AUC∞. Odpowiednie 90% przedziały ufności dla stosunku parametrów farmakokinetycznych po 6 miesiącach badania w porównaniu z wartościami wyjściowymi mieściły się w granicach okna ekwiwalentności (od 80% do 125%). Wskazuje to na brak zależnych od czasu zmian w właściwościach farmakokinetycznych Refaktor AF.
W tym samym badaniu, w którym aktywność leku Refaktor AF, rekombinowanego czynnika VIII o pełnej długości cząsteczki (lek porównawczy) oraz aktywność czynnika VIII oznaczano w próbkach osocza pacjentów (30 pacjentów, którzy wcześniej otrzymywali leczenie, ≥ 12 lat) metodą jednoetapowego testu krzepnięcia krwi, Refaktor AF wykazał farmakokinetyczną bioekwiwalentność w porównaniu z lekiem porównawczym przy zastosowaniu standardowego podejścia do ustalenia bioekwiwalentności.
U pacjentów, którzy wcześniej nie otrzymywali leczenia, parametry farmakokinetyczne Refaktor oceniano metodą analizy chromogennej. U tych pacjentów (n = 59; mediana wieku 10 ± 8,3 miesiąca) średnie odzyskanie w tygodniu „0” wynosiło 1,5 ± 0,6 j.m./dl na j.m./kg (w zakresie 0,2–2,8 j.m./dl na j.m./kg), co było niższe niż wartości uzyskane u pacjentów, którzy wcześniej otrzymywali leczenie, w tygodniu „0” ze średnią wartością odzyskania 2,4 ± 0,4 j.m./dl na j.m./kg (w zakresie 1,1–3,8 j.m./dl na j.m./kg). U pacjentów, którzy wcześniej nie otrzymywali leczenia, średnie odzyskanie było stabilne przez okres 2-letni (5 wizyt w tym okresie) i mieściło się w granicach 1,5–1,8 j.m./dl na j.m./kg. Badanie z wykorzystaniem populacyjnego modelu farmakokinetycznego, obejmujące dane 44 pacjentów, którzy wcześniej nie otrzymywali leczenia, wykazało, że przewidywany średni okres półtrwania wynosi 8,0 ± 2,2 godziny.
W badaniu zastosowania leku Refaktor AF wśród 19 PNP, poziom odzysku u 17 dzieci w wieku od 28 dni do 2 lat wynosił 1,32 ± 0,65 j.m./dl na j.m./kg, a u 2 dzieci w wieku od 2 do < 6 lat – odpowiednio 1,7 i 1,8 j.m./dl na j.m./kg. Z wyjątkiem przypadków, w których wykryto inhibitory, średni poziom odzysku był stabilny w czasie (6 wizyt w okresie 2-letnim), a pojedyncze wartości wahały się od 0 (w obecności inhibitora) do 2,7 j.m./dl na j.m./kg. W tabeli 3 przedstawiono zaobserwowane parametry farmakokinetyczne Refaktor AF po podaniu dawki 50 j.m./kg w badaniu przeprowadzonym wśród 37 wcześniej leczonych dzieci.
Tabela 3
| Średnie wartości ± SD parametrów farmakokinetycznych czynnika VIII po podaniu jednej dawki 50 J/ kg u dzieci (PLP) |
||
| Parametr |
Liczba podmiotów |
Średnia wartośća ± SD |
| Odzysk, J/dl na J/kg W wieku do 6 lat W wieku od 6 do 12 lat |
17 19 |
1,7 ± 0,4 2,1 ± 0,8 |
| Cmax, J/mlb |
19 |
0,9 (45) |
| AUCinf, J∙godz/mlb |
14 |
9,9 (41) |
| t½, godzinyb |
14 |
9,1 ± 1,9 |
| CL, ml/godz/kgb |
14 |
4,4 (30) |
| Vss, ml/kgb |
14 |
56,4 (15) |
| a Średnia wartość geometryczna (geometryczny %WSD) we wszystkich przypadkach, z wyjątkiem średniej arytmetycznej ± SD dla odzysku przyrostowego oraz t½. b Tylko pacjenci w wieku od 6 do 12 lat. Skróty: Cmax – maksymalna stężenie w osoczu; WSD – współczynnik zmienności; AUCinf – pole pod krzywą „stężenie w osoczu – czas” od 0 do nieskończoności; t½ – okres półtrwania; CL – klirens; SD – odchylenie standardowe; Vss – objętość rozłożenia w stanie stacjonarnym. |
||
Dane kliniczne.
Wskazania.
Leczenie i zapobieganie krwawieniom u pacjentów z hemofilią A (wrodzony niedobór czynnika krzepnięcia krwi VIII).
Refaktor AF wskazany jest do stosowania u dorosłych i dzieci w każdym wieku, w tym u niemowląt.
Refaktor AF nie zawiera czynnika von Willebranda i dlatego nie jest wskazany w leczeniu choroby von Willebranda.
Przeciwwskazania.
Nadwrażliwość na substancję czynną lub na którąkolwiek z substancji pomocniczych.
Znana reakcja alergiczna na białka chomika.
Interakcje z innymi lekami i inne rodzaje interakcji.
Nie zgłaszano interakcji rekombinowanego czynnika krzepnięcia krwi ludzkiej VIII z innymi lekami.
Szczególne środki ostrożności.
Śledzenie
W celu poprawy śledzenia produktów biologicznych należy dokładnie rejestrować nazwę handlową oraz numer serii zastosowanego leku. W tym celu pacjenci mogą przykleić jeden z odklejanych samoprzylepnych nalepek z butelki lub wstępnie wypełnionego strzykawki do swojego dziennika, aby udokumentować numer serii, lub wykorzystać go do zgłoszenia wszelkich działań niepożądanych.
Podatność na reakcje nadwrażliwościowe
Refaktor AF może powodować rozwój reakcji nadwrażliwościowych typu alergicznego. Preparat zawiera niewielkie ilości białek chomika. Pacjentów należy poinstruować, że w przypadku wystąpienia objawów nadwrażliwości należy natychmiast przerwać stosowanie leku i skontaktować się z lekarzem. Pacjenci powinni być poinformowani o wczesnych objawach reakcji nadwrażliwościowych, w tym o pokrzywce, uogólnionej pokrzywce, uczuciu ucisku w klatce piersiowej, świstach w oddychaniu, nadciśnieniu tętniczym oraz anafilaksji.
W przypadku wystąpienia wstrząsu należy podjąć standardowe leczenie stanu wstrząsu.
Inhibitory
Wytwarzanie neutralizujących przeciwciał (inhibitorów) czynnika VIII jest znanym powikłaniem występującym podczas leczenia osób z hemofilią typu A. Zazwyczaj są to immunoglobuliny IgG działające przeciwko aktywności prokoagulacyjnej czynnika VIII. Ich obecność w osoczu oznacza się metodą ilościową zmodyfikowaną i wyraża w jednostkach Bethesda (JB) na 1 ml. Ryzyko powstawania inhibitorów koreluje z ciężkością choroby oraz z ekspozycją na czynnik VIII, przy czym jest najwyższe w ciągu pierwszych 50 dni stosowania leku, ale utrzymuje się przez całe życie, choć występuje rzadko.
Kliniczne znaczenie powstawania inhibitorów zależy od ich miana – inhibitory o niskim mianie stanowią mniejsze ryzyko niewystarczającej odpowiedzi klinicznej niż inhibitory o wysokim mianie.
Należy dokładnie monitorować pacjentów otrzymujących leczenie rekombinowanymi czynnikami krzepnięcia krwi VIII pod kątem powstawania inhibitorów, stosując odpowiednią obserwację kliniczną i badania laboratoryjne. Jeśli oczekiwane poziomy aktywności czynnika VIII w osoczu krwi nie zostaną osiągnięte lub jeśli krwawienie nie ustaje pomimo zastosowania odpowiedniej dawki leku, należy przeprowadzić badania w celu wykrycia obecności inhibitorów czynnika VIII. U pacjentów z wysokimi stężeniami inhibitorów terapia czynnikiem VIII może być nieskuteczna, należy wówczas rozważyć możliwość zastosowania innych leków. Leczenie takich pacjentów powinno być prowadzone przez lekarzy doświadczonych w terapii hemofilii oraz w przypadkach powstawania inhibitorów czynnika VIII.
Zgłaszanie niewystarczającej skuteczności
W badaniach klinicznych oraz w okresie po wprowadzeniu na rynek Refaktor AF zgłaszano przypadki niewystarczającej skuteczności leku, głównie u pacjentów stosujących lek w celu profilaktyki. Brak skuteczności opisywano jako krwawienia do sędziwych stawów, krwawienia do nowych stawów lub subiektywne odczucie przez pacjenta początku nowego krwawienia. Przy przepisywaniu Refaktor AF ważne jest indywidualne dobrać dawkowanie oraz monitorować poziom czynnika u każdego pacjenta w celu zapewnienia odpowiedniej odpowiedzi terapeutycznej (patrz sekcja „Działania niepożądane”).
Powikłania sercowo-naczyniowe
U pacjentów z istniejącymi czynnikami ryzyka rozwoju powikłań sercowo-naczyniowych leczenie zastępcze czynnikiem VIII może prowadzić do zwiększenia ryzyka kardiowaskularnego.
Powikłania związane z cewnikiem
W przypadku konieczności stosowania urządzenia do dostępu do żyły centralnej należy wziąć pod uwagę ryzyko powikłań związanych z jego użyciem, w tym infekcji miejscowych, bakteriemii oraz zakrzepicy cewnika (patrz sekcja „Działania niepożądane”).
Zawartość sodu
Po rozpuszczeniu proszku 1 butelka lub wstępnie wypełniona strzykawka zawiera 1,27 mmol (lub 29 mg) sodu, co odpowiada 1,5% zalecanej przez WHO maksymalnej dziennej dawki sodu dla dorosłego człowieka wynoszącej 2 g. W zależności od masy ciała pacjenta oraz dawki Refaktor AF pacjenci mogą stosować kilka butelek lub wstępnie wypełnionych strzykawek. Informację tę należy uwzględnić u pacjentów przestrzegających diety ograniczającej spożycie sodu.
Stosowanie w czasie ciąży lub karmienia piersią.
Nie przeprowadzono badań wpływu czynnika VIII na funkcję rozrodczą u zwierząt, dlatego brakuje danych dotyczących jego wpływu na płodność. Ponieważ hemofilia typu A rzadko występuje u kobiet, brakuje doświadczenia w stosowaniu czynnika VIII w czasie ciąży i karmienia piersią. Dlatego czynnik VIII należy stosować w czasie ciąży i karmienia piersią wyłącznie w razie nagłej potrzeby.
Wpływ na zdolność prowadzenia pojazdów lub obsługiwania maszyn.
Refaktor AF nie wpływa na zdolność prowadzenia pojazdów oraz pracy z innymi maszynami.
Sposób stosowania i dawki.
Leczenie należy rozpoczynać pod nadzorem lekarza z doświadczeniem w leczeniu hemofilii A.
Kontrola leczenia.
W celu ustalenia dawki wymaganej w trakcie leczenia oraz częstotliwości powtarzanych infuzji zaleca się przeprowadzanie badań poziomu czynnika VIII. U różnych pacjentów odpowiedź na podanie czynnika VIII może się różnić, co objawia się różnymi poziomami odbudowy oraz różnymi okresami półwydalenia. Dawkę obliczoną według masy ciała pacjenta może być konieczne skorygować u chorych z nadmierną lub niedostateczną masą ciała. W szczególności w przypadku dużych zabiegów chirurgicznych konieczne jest staranne monitorowanie terapii zastępczej za pomocą analizy krzepnięcia (aktywności czynnika VIII w osoczu).
W celu monitorowania poziomu aktywności czynnika VIII u pacjentów podczas leczenia preparatem Refaktor AF zaleca się stosowanie metody analizy chromogennej. Jeśli do oznaczania aktywności czynnika VIII w próbkach krwi pacjentów stosuje się in vitro aktywowany częściowy czas tromboplastynowy (APTT), obliczany za pomocą jednoetapowego testu aktywności krzepnięcia, wyniki aktywności czynnika VIII mogą być wpływane zarówno przez rodzaje odczynników, jak i przez stosowane wzorce standardowe. Mogą również występować istotne różnice między wynikami oznaczeń ilościowych uzyskanymi metodą analizy chromogennej a oznaczeniami APTT za pomocą jednoetapowego testu aktywności krzepnięcia. Zazwyczaj wyniki uzyskane metodą jednoetapowego testu aktywności krzepnięcia są o 20–50% niższe niż wyniki oznaczeń ilościowych wykonanych metodą analizy chromogennej. Do skorygowania tej rozbieżności można stosować laboratoryjny standard Refaktor AF (patrz sekcja „Farmakokinetyka”). Jest to szczególnie ważne, gdy stosowane są inne laboratoria i/lub odczynniki.
Dozowanie.
Dawkowanie i trwanie terapii zastępczej zależy od stopnia niedoboru czynnika VIII, lokalizacji i nasilenia krwawienia oraz od stanu klinicznego pacjenta. Dawkowanie należy dostosować do odpowiedzi klinicznej pacjenta. W przypadku obecności inhibitorów może być konieczne zastosowanie wyższych dawek leku lub odpowiedniego specyficznego leczenia.
Ilość jednostek czynnika VIII podaje się w j.m. (jednostkach międzynarodowych) powiązanych z działającym standardem WHO dla leków zawierających czynnik VIII. Aktywność czynnika VIII we krwi osoczu podaje się w procentach (w stosunku do jego zawartości w normalnym ludzkim osoczu) lub w jednostkach międzynarodowych (w stosunku do międzynarodowego standardu czynnika VIII w osoczu). 1 j.m. aktywności czynnika VIII odpowiada ilości czynnika VIII w 1 ml normalnego ludzkiego osoczu.
Aktywność innego preparatu moroctokogu alfa (Xyntha), zarejestrowanego poza Ukrainą, określa się za pomocą standardu aktywności produkcyjnej, który został skalibrowany w odniesieniu do międzynarodowego standardu WHO przy użyciu jednoetapowego testu aktywności krzepnięcia. Ze względu na różnice między metodami stosowanymi do oznaczania aktywności preparatów Xyntha i Refaktor AF, 1 j.m. preparatu Xyntha (skalibrowanego metodą jednoetapową) odpowiada w przybliżeniu 1,38 j.m. preparatu Refaktor AF (skalibrowanego metodą chromogenną). Jeśli pacjentowi, który zazwyczaj otrzymuje leczenie preparatem Xyntha, przepisze się Refaktor AF, lekarz może rozważyć możliwość skorygowania dawki w zależności od wskaźników odbudowy poziomu czynnika VIII.
Pacjentom z hemofilią A zaleca się noszenie przy sobie wystarczającej ilości preparatu czynnika VIII zgodnie z obowiązującym schematem leczenia na wypadek potrzeby leczenia podczas podróży. Pacjentów należy zachęcać do konsultacji z lekarzem przed podróżą.
Leczenie na żądanie.
Obliczenie wymaganej dawki czynnika VIII opiera się na danych empirycznych, że 1 j.m. czynnika VIII na 1 kg masy ciała zwiększa aktywność czynnika VIII we krwi osoczu o 2 j.m./dl. Wymaganą dawkę oblicza się według wzoru:
wymagana liczba jednostek (j.m.) =
masa ciała (kg) × pożądane zwiększenie ilości czynnika VIII (%, lub j.m./dl) × 0,5 (j.m./kg na j.m./dl),
gdzie 0,5 j.m./kg na j.m./dl jest wartością odwrotną do odbudowy, która zazwyczaj występuje po infuzji czynnika VIII.
Dawkę przepisanego preparatu oraz częstotliwość stosowania należy zawsze ustalać indywidualnie, uwzględniając skuteczność kliniczną leku.
W przypadku wystąpienia objawów krwawienia aktywność czynnika VIII we krwi osoczu nie powinna spadać poniżej poziomów (w % lub w j.m./dl) wskazanych w tabeli 4. Tabelę tę można wykorzystać jako wskazówkę dotyczącą dawkowania czynnika VIII w przypadku krwawień i zabiegów chirurgicznych.
Tabela 4
| Ciężkość krwawienia/ typ zabiegu chirurgicznego |
Wymagany poziom czynnika VIII (% normy lub j.m./dcl osocza) |
Częstotliwość podawania (godziny)/ trwanie terapii (dni) |
| Krwawienia |
||
| Początkowe objawy hemartrozy, krwawienia do mięśni lub krwawienia z jamy ustnej |
20–40 |
Powtarzać podawanie co 12–24 godziny przez co najmniej 1 dzień do ustania krwawienia, o czym świadczy brak bólu lub obecność gojenia |
| Średnio nasilona hemartroza, krwawienia do mięśni lub siniaki |
30–60 |
Powtarzać podawanie co 12–24 godziny przez 3–4 dni lub dłużej, aż do zniknięcia bólu i przywrócenia ruchomości kończyn |
| Krwawienia zagrożone dla życia |
60–100 |
Powtarzać podawanie co 8–24 godziny do ustąpienia zagrożenia dla życia |
| Zabiegi chirurgiczne |
||
| Niewielkie zabiegi chirurgiczne, w tym ekstrakcja zęba |
30–60 |
Powtarzać podawanie co 24 godziny przez co najmniej 1 dzień do gojenia |
| Uciążliwe zabiegi chirurgiczne |
80–100 (przed i po zabiegu chirurgicznym) |
Powtarzać podawanie co 8–24 godziny do skutecznego gojenia rany, następnie kontynuować terapię przez co najmniej 7 dni w celu utrzymania aktywności czynnika VIII na poziomie 30–60 % (j.m./dcl) |
Profilaktyka
Długoterminową profilaktykę krwawień u pacjentów z ciężką hemofilią typu A prowadzi się zazwyczaj stosując 20–40 MI czynnika VIII na 1 kg masy ciała co 2–3 dni. W niektórych przypadkach, szczególnie u młodszych pacjentów, może być konieczne zwiększenie dawki lub częstotliwości podawania leku.
Dzieci
U dzieci w młodszym wieku (do 6 roku życia) leczonych lekiem Refaktor AF może być konieczne zastosowanie wyższych dawek w porównaniu z dawkami stosowanymi u dorosłych pacjentów i dzieci w starszym wieku (patrz sekcja „Farmakokinetyka”).
Pacjenci w wieku podeszłym
Pacjenci powyżej 65 roku życia nie byli objęci badaniami klinicznymi. Dawkę dla pacjentów w wieku podeszłym należy dobierać indywidualnie.
Pacjenci z niewydolnością nerek lub wątroby
Dostosowanie dawki u pacjentów z niewydolnością nerek lub wątroby nie było badane w trakcie badań klinicznych.
Sposób stosowania.
Dożylne.
Refaktor AF podaje się dożylnie w postaci wlewu przez kilka minut po rozpuszczeniu w odpowiednim rozpuszczalniku. Szybkość podania należy dostosować do komfortu pacjenta. Osobom samodzielnie podającym lek, ale niebędącym pracownikami medycznymi, zaleca się odpowiednie przeszkolenie.
Postać – fiolka
Liofilizat rozpuszcza się w odpowiednim rozpuszczalniku (roztwór chlorku sodu 0,9 %) dostarczanym w szprycie wstępnie napełnionym, przy użyciu sterylnego adaptera. Po dodaniu rozpuszczalnika fiolkę należy ostrożnie obracać aż do całkowitego rozpuszczenia się proszku.
Szczegółowe informacje dotyczące przygotowania i stosowania roztworu podano poniżej.
Po rozpuszczeniu roztwór ponownie pobiera się do szpryca. Otrzymany roztwór powinien być klarowny lub lekko opalescencyjny i bezbarwny. Należy odrzucić roztwór, jeśli obserwuje się obecność zanieczyszczeń lub zmianę barwy.
Postać – wstępnie napełniony szpryc:
Liofilizowany proszek w górnej komorze wstępnie napełnionego szpryca należy rozpuścić za pomocą rozpuszczalnika (roztwór chlorku sodu, 9 mg/ml (0,9 %)), znajdującego się w dolnej komorze. Należy ostrożnie obracać wstępnie napełniony szpryc, aż do całkowitego rozpuszczenia się proszku. Szczegółowe informacje dotyczące przygotowania i stosowania roztworu podano poniżej.
Po odtworzeniu roztwór będzie klarowny lub lekko opalescencyjny i bezbarwny. Należy odrzucić roztwór, jeśli widoczne są mechaniczne zanieczyszczenia lub zmiana barwy.
Lek Refaktor AF po rozpuszczeniu zawiera polisorbat 80, który może przyspieszać ekstrakcję di-(2-etyloheksylu) ftalanu z poli(chlorku winylu) (PVC). Należy wziąć pod uwagę tę właściwość przy przygotowaniu i stosowaniu leku, w tym czas przechowywania w pojemniku z PVC po przygotowaniu roztworu. Ważne jest dokładne przestrzeganie zaleceń podanych w sekcji „Warunki przechowywania”.
Nieużywane resztki leku lub materiały używane do rozpuszczania i podawania leku należy zutylizować zgodnie z lokalnymi przepisami.
Postać – fiolka
Przygotowanie roztworu:
- Doprowadź temperaturę liofilizatu i rozpuszczalnika w szprycie wstępnie napełnionym do temperatury pokojowej.
- Zdejmij plastikowy korek typu flip-top z fiolki Refaktor AF, aby odsłonić środkową część gumowej zawleczki.
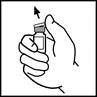
- Przetrzyj górną część fiolki tamponem alkoholowym dostarczonym w opakowaniu lub użyj innego środka antyseptycznego i pozwól wyschnąć. Po oczyszczeniu nie dotykaj rękami gumowej zawleczki i nie dopuszczaj, aby dotykała ona żadnych powierzchni.
- Odsuń osłonę ochronną przezroczystej plastikowej opakowania adaptera do fiolki. Nie wyciągaj adaptera z opakowania.
- Postaw fiolkę na płaskiej powierzchni. Trzymając opakowanie z adapterem, załóż adapter na fiolkę. Silnie naciśnij, aż adapter zatrzasnie się na górnym końcu fiolki, a jego ostrze przejdzie przez zawleczkę fiolki.
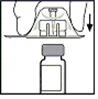
- Zdejmij opakowanie z adaptera i zutylizuj je.
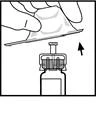
- Przyłącz tłok strzykawki do szpryca z rozpuszczalnikiem: włóż tłok do otworu w zatyczce strzykawki, silnie naciskaj i obracaj tłok, aż będzie pewnie osadzony w zatyczce.
- Odlamaj plastikowy kaptur z wskaźnikiem pierwszego otwarcia ze strzykawki z rozpuszczalnikiem, niszcząc integralność perforacji na kapturze. Należy to zrobić, kołysząc kaptur, aż perforacja zostanie złamana. Nie dotykaj wewnętrznej powierzchni kaptura ani końcówki strzykawki. Może być konieczne ponowne założenie kaptura (jeśli przygotowany Refaktor AF nie zostanie użyty natychmiast), dlatego odłóż go, ustawiając otworem do góry.
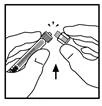
- Postaw fiolkę na płaskiej powierzchni. Przyłącz strzykawkę z rozpuszczalnikiem do adaptera fiolki, wsuwając końcówkę strzykawki do otworu adaptera, silnie naciskając i obracając strzykawkę zgodnie z ruchem wskazówek zegara, aż do uzyskania pewnego połączenia.
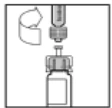
- Powoli naciśnij tłok strzykawki, aby wprowadzić cały rozpuszczalnik do fiolki Refaktor AF.
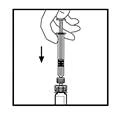
- Zachowując strzykawkę przyłączoną do adaptera, ostrożnie obracaj fiolkę aż do całkowitego rozpuszczenia się proszku.
- Przed zastosowaniem przygotowany roztwór należy wizualnie sprawdzić pod kątem obecności zanieczyszczeń. Otrzymany roztwór powinien być klarowny lub lekko opalescencyjny i bezbarwny.
Jeśli do jednej infuzji stosuje się więcej niż jedną fiolkę leku Refaktor AF, każdą fiolkę należy przygotować zgodnie z powyższymi instrukcjami. Następnie należy usunąć strzykawkę z rozpuszczalnikiem, pozostawiając adapter do fiolki na miejscu, a do pobrania roztworu z każdej fiolki można użyć jednej dużej strzykawki z zatrzaskiem Luer.
- Upewniwszy się, że tłok pozostaje całkowicie włożony do cylindra strzykawki, odwróć fiolkę. Powoli wyciągnij cały roztwór przez adapter fiolki do strzykawki.
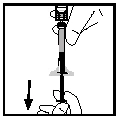
- Odłącz strzykawkę od adaptera fiolki, ostrożnie odciągając i obracając strzykawkę przeciwnie do ruchu wskazówek zegara. Wyrzuć fiolkę z przyłączonym adapterem.
Jeśli roztwór nie zostanie użyty natychmiast, kaptur strzykawki należy ostrożnie ponownie założyć. Nie dotykaj końcówki strzykawki ani wewnętrznej powierzchni kaptura.
Przygotowany roztwór należy użyć natychmiast lub w ciągu 3 godzin od przygotowania. Przed zastosowaniem przygotowany roztwór można przechowywać w temperaturze nie wyższej niż 25 °C.
Zastosowanie (infuzja dożylna)
Refaktor AF należy podawać, używając dostarczonego zestawu do infuzji oraz wstępnie napełnionego rozpuszczalnikiem strzykawki dostarczonego w zestawie lub jednej sterylnego jednorazowego strzykawki plastikowej z zatrzaskiem Luer.
- Przyłącz strzykawkę do złącza Luer zestawu do infuzji.
- Załóż opaskę uciskową i przygotuj miejsce wstrzyknięcia, dokładnie przetrzyj skórę tamponem alkoholowym dostarczonym w zestawie.
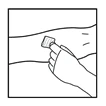
- Wprowadź igłę zestawu do infuzji do żyły i zdjąć opaskę uciskową. Usuń powietrze z zestawu do infuzji, odciągając strzykawkę. Przygotowany lek należy podawać dożylnie przez kilka minut. Lekarz może dostosować zalecaną szybkość infuzji, aby procedura była bardziej komfortowa dla pacjenta.
Postać – wstępnie napełniony szpryc
Przygotowanie roztworu:
- Doprowadź wstępnie napełniony szpryc do temperatury pokojowej.
- Wyjmij zestaw do wstępnie napełnionego szpryca Refaktor AF i połóż na czystej powierzchni, upewniając się, że masz wszystkie niezbędne elementy.
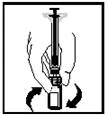
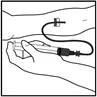
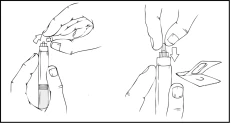
- Trzymaj tłok strzykawki tak, jak pokazano na poniższym schemacie. Mocno wkręć tłok strzykawki w otwór w podstawce dla palców wstępnie napełnionego szpryca Refaktor AF, naciskając i obracając zgodnie z ruchem wskazówek zegara, aż poczujesz opór (około 2 obroty).
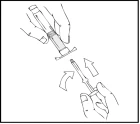
Podczas całego procesu przygotowania roztworu należy trzymać wstępnie napełniony szpryc Refaktor AF pionowo (biały proszek powinien być nad przezroczystym roztworem), aby uniknąć możliwego wycieku.
- Trzymając wstępnie napełniony szpryc w pozycji pionowej, odlamaj biały ochronny kaptur z wskaźnikiem pierwszego otwarcia, odginając go z prawej do lewej (lub delikatnymi ruchami kołyszącymi), aby złamać perforację kaptura i zobaczyć szary gumowy kaptur końcówki wstępnie napełnionego szpryca Refaktor AF.
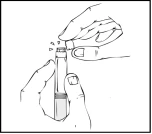
- Wyjmij z opakowania niebieski ochronny wentylowany sterylny kaptur.
Trzymając wstępnie napełniony szpryc Refaktor AF cały czas w pozycji pionowej, usuń szary gumowy kaptur i zastąp go ochronnym niebieskim wentylowanym kaptrem. Ten ochronny kaptur ma małe otwory umożliwiające wyjście powietrza, co zapobiega nadmiernemu ciśnieniu. Nie dotykaj odkrytego końca strzykawki ani niebieskiego ochronnego kaptura.
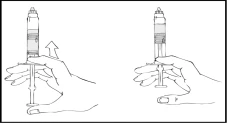
- Ostrożnie i powoli przesuwaj tłok strzykawki, naciskając aż do momentu, gdy dwa tłoki wewnątrz wstępnie napełnionego szpryca spotkają się i cały rozpuszczalnik przejdzie do górnej komory zawierającej proszek Refaktor AF.
Uwaga: Aby zapobiec wyciekowi cieczy z końcówki strzykawki, nie naciskaj tłoka strzykawki z nadmierną siłą.
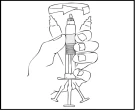
- Trzymając szpryc Refaktor AF w pozycji pionowej, ostrożnie obracaj go aż do rozpuszczenia się proszku.
Wizualnie sprawdź roztwór pod kątem obecności zanieczyszczeń lub zmiany koloru. Roztwór powinien być klarowny, lekko opalescencyjny i bezbarwny. Jeśli roztwór w wstępnie napełnionym szprycie zawiera zanieczyszczenia lub zmienił barwę, nie należy go używać.
- Kontynuując trzymanie wstępnie napełnionego szpryca Refaktor AF w pozycji pionowej, powoli przesuwaj tłok strzykawki, aż większość, ale nie cały, powietrza zostanie usunięte z (górnej) komory.
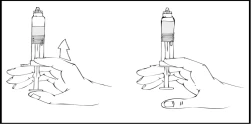
Refaktor AF należy podawać w ciągu 3 godzin od przygotowania roztworu lub usunięcia szarego gumowego kaptura z wstępnie napełnionego szpryca.
Jeśli nie będziecie natychmiast używać roztworu Refaktor AF, należy przechowywać szpryc w pozycji pionowej z niebieskim ochronnym kaptrem na wstępnie napełnionym szprycu, aż będziecie gotowi do przeprowadzenia infuzji. Przygotowany roztwór może być przechowywany w temperaturze pokojowej przez 3 godziny. Jeśli nie zostanie użyty w ciągu 3 godzin, roztwór należy wyrzucić.
Zastosowanie (infuzja dożylna)
Refaktor AF należy podawać, używając dostarczonego zestawu do infuzji.
- Zdejmij ochronny niebieski kaptur i pewnie przyłącz zestaw do infuzji do wstępnie napełnionego szpryca Refaktor AF.
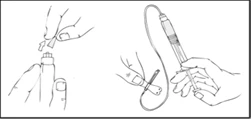
- Załóż opaskę uciskową i przygotuj miejsce wstrzyknięcia, dokładnie przetrzyj skórę tamponem alkoholowym dostarczonym w zestawie.
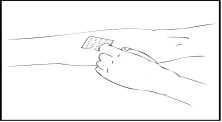
- Zdejmij ochronny kaptur igły i wprowadź igłę zestawu do infuzji do żyły. Zdejmij opaskę uciskową. Przygotowany roztwór leku Refaktor AF należy podawać dożylnie przez kilka minut. Lekarz może dostosować zalecaną szybkość infuzji, aby procedura była bardziej komfortowa dla Ciebie. Nie próbuj samodzielnie przeprowadzać infuzji bez odpowiedniego przygotowania.
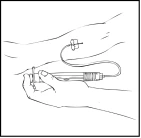
Przygotowanego roztworu Refaktor AF nie można podawać przez ten sam zestaw do infuzji lub pojemnik z innymi lekami.
- Po infuzji Refaktor AF usuń zestaw do infuzji i wyrzuć go. Ilość leku pozostająca w zestawie do infuzji nie wpływa na leczenie. Dodatkowe informacje dotyczące stosowania kilku szpryców Refaktor AF z 10 cm³ lub większymi szprycami typu Luer Lock (szprycy 10 cm³ lub większe szprycy typu Luer Lock nie są częścią opakowania)
- Przygotuj roztwór we wszystkich szprycach Refaktor AF zgodnie z instrukcjami przygotowania roztworu podanymi powyżej.
Trzymając napełniony szpryc Refaktor AF w pozycji pionowej, powoli przesuwaj tłok strzykawki, aż większość, ale nie cały, powietrza zostanie usunięte ze strzykawki.
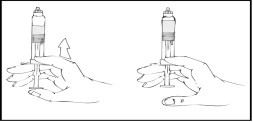
- Otwórz połączenie do szpryców typu Luer Lock (nie zawarte w opakowaniu).
- Podłącz sterylny szpryc 10 cm³ lub większy typu Luer Lock do jednego otworu (portu) w połączeniu do szpryców, a szpryc Refaktor AF do drugiego otwartego portu po przeciwnej stronie.
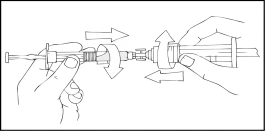
- Trzymając szpryc Refaktor AF u góry, powoli naciskaj tłok strzykawki, aż zawartość przejdzie do pustego szpryca 10 cm³ lub większego typu Luer Lock.
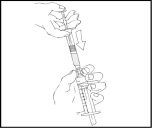
- Usuń pusty wstępnie napełniony szpryc Refaktor AF i powtarzaj procedury 3 i 4 z dodatkowymi szprycami z przygotowanym roztworem.
- Usuń połączenie do szpryców z szpryca 10 cm³ lub większego typu Luer Lock i przyłącz zestaw do infuzji, jak opisano powyżej w instrukcji podania.
Nieużywany roztwór, pustą fiolkę(-ki) oraz używane igły i szprycy należy zutylizować w odpowiednim pojemniku do utylizacji odpadów medycznych, ponieważ mogą one zaszkodzić innym, jeśli nie zostaną odpowiednio zutylizowane.
Dzieci.
Refaktor AF można stosować dzieciom w każdym wieku, w tym niemowlętom, zgodnie z informacjami w sekcji „Sposób stosowania i dawki”.
Przedawkowanie.
Nie odnotowano żadnych objawów przedawkowania przy stosowaniu leków rekombinowanego czynnika VIII.
Działania niepożądane.
Podsumowanie profilu bezpieczeństwa
Reakcje nadwrażliwościowe i alergiczne (które mogą obejmować obrzęk naczynioruchowy, uczucie pieczenia i mrowienia w miejscu infuzji, dreszcze, rumień, uogólnione pokrzywiczkę, ból głowy, pokrzywkę, hipotensję tętniczą, letargię, nudności, pobudzenie, tachykardię, uczucie ucisku w klatce piersiowej, szum w uszach, wymioty, świsty podczas oddychania) po zastosowaniu Refaktor AF występują rzadko i w niektórych przypadkach mogą postępować do ciężkiej postaci anafilaksji, w tym do wstrząsu (patrz sekcja „Szczególne środki ostrożności”).
Refaktor AF może zawierać śladowe ilości białka chomika. Bardzo rzadko obserwowano powstawanie przeciwciał przeciwko białku chomika, jednak nie stwierdzono powikłań klinicznych. W badaniu z udziałem Refaktor, u 20 z 113 (18%) pacjentów wcześniej leczonych (PPL) zaobserwowano wzrost miana przeciwciał przeciwko białku jajników chińskiego chomika, bez widocznego wpływu klinicznego.
Powstawanie przeciwciał neutralizujących (inhibitorów) może występować u pacjentów z hemofilią A leczonych czynnikiem VIII, w tym Refaktor AF. Powstawanie inhibitorów może objawiać się niewystarczającą odpowiedzią kliniczną. W takich przypadkach zaleca się skonsultowanie się z wyspecjalizowanym ośrodkiem leczenia hemofilii.
Lista działań niepożądanych
Informacja zamieszczona poniżej została sklasyfikowana według systemu organów zgodnie z Medycznym Słownikiem do Celów Regulacyjnych (MedDRA). Działania niepożądane sklasyfikowano według częstości występowania: bardzo często (≥ 1/10); często (od ≥ 1/100 do < 1/10) oraz rzadko (od ≥ 1/1000 do < 1/100). Poniżej przedstawiono działania niepożądane obserwowane w badaniach klinicznych z udziałem Refaktor lub Refaktor AF. Częstość odnosi się do wszystkich przypadkowych działań niepożądanych, które wystąpiły podczas stosowania leku we wszystkich badaniach klinicznych z udziałem 715 pacjentów.
W każdej grupie działania niepożądane wymieniono w kolejności malejącej według ich nasilenia.
Z układy krwiotwórczego i limfatycznego:
bardzo często: inhibicja czynnika VIII (u pacjentów wcześniej nieleczonych)*;
rzadko: inhibicja czynnika VIII (u pacjentów wcześniej leczonych)*.
Z układy odpornościowego:
rzadko: reakcje anafilaktyczne.
Zaburzenia metaboliczne i zaburzenia odżywiania:
często: zmniejszenie apetytu.
Z układy nerwowego:
bardzo często: ból głowy;
często: zawroty głowy;
rzadko: neuropatia obwodowa, senność, dysgezja.
Z układy serca:
rzadko: dławica, tachykardia, kołatanie serca.
Z układy naczyń:
często: krwawienie/krwawienie podskórne;
rzadko: hipotensja, zapalenie żył z torem zakrzepowym, zaczerwienienie twarzy.
Z układy oddechowego, narządów klatki piersiowej i śródpiersia:
bardzo często: kaszel;
rzadko: duszność.
Z układy pokarmowego:
często: biegunka, nudności, ból brzucha, wymioty.
Z układy skóry i tkanek podskórnych:
często: pokrzywka, swędzenie, wysypka;
rzadko: hiperhidroza.
Z układy mięśniowo-szkieletowego i tkanki łącznej:
bardzo często: artrodynia;
często: mialgia.
Ogólne zaburzenia i reakcje w miejscu podania:
bardzo często: podwyższenie temperatury ciała;
często: dreszcze, powikłania związane z trwałym cewnikiem dożylnym;
rzadko: osłabienie, ból, stan zapalny i inne reakcje w miejscu podania.
Badania laboratoryjne:
często: dodatni test na przeciwciała; dodatni test na przeciwciała przeciwko czynnikowi VIII;
rzadko: wzrost stężenia asparaginianaminotransferazy, alaninianaminotransferazy, bilirubiny we krwi, kreatynofosfokinazy we krwi.
* Częstość podana na podstawie badań wszystkich czynników VIII, w których uczestniczyli pacjenci z ciężką hemofilią A. PPL — pacjenci wcześniej leczeni; PPN — pacjenci wcześniej nieleczoni.
Dzieci
Zgłoszono jeden przypadek powstania torbieli u 11-letniego pacjenta oraz jeden przypadek omdlenia u 13-letniego pacjenta; rozwój tych stanów może być związany z leczeniem lekiem Refaktor AF.
Bezpieczeństwo stosowania Refaktor AF oceniano w badaniach z udziałem dorosłych, dzieci i nastolatków wcześniej leczonych (n = 18, w wieku 12–16 lat w badaniu oraz n = 49, w wieku 7–16 lat w dodatkowym badaniu) i stwierdzono tendencję do wzrostu częstości niepożądanych zdarzeń u dzieci w wieku 7–16 lat w porównaniu z dorosłymi.
Dodatkowe dane dotyczące bezpieczeństwa uzyskano w badaniach z udziałem pacjentów wcześniej leczonych (n = 18 w wieku do 6 lat oraz n = 19 w wieku 7–12 lat) lub wcześniej nieleczonych (n = 23 w wieku do 6 lat). Uzyskane dane wskazują na podobieństwo profilu bezpieczeństwa do profilu u dorosłych pacjentów.
Zgłaszanie podejrzewanych działań niepożądanych.
Istotne jest przekazywanie informacji o podejrzewanych działaniach niepożądanych po rejestracji leku. Pozwala to na dalsze monitorowanie stosunku korzyści do ryzyka stosowania leku. Osoby sprawujące zawód medyczny powinny zgłaszać wszystkie podejrzewane działania niepożądane zgodnie z lokalnymi wymaganiami.
Okres ważności.
Postać opakowania – fiolka: dla proszku – 3 lata, dla rozpuszczalnika – 5 lat.
Postać opakowania – szpryt wstępnie napełniony: 3 lata.
Warunki przechowywania.
Przechowywać w oryginalnym opakowaniu w temperaturze od 2 do 8 °C. Nie zamrażać, aby uniknąć uszkodzenia szpryta wstępnie napełnionego.
Lek w okresie ważności może być przechowywany w temperaturze nie wyższej niż 25 °C przez okres do 3 miesięcy. Leku nie można ponownie umieszczać w lodówce, jeśli był przechowywany w temperaturze pokojowej.
Przygotowany roztwór należy użyć natychmiast lub w ciągu 3 godzin od przygotowania roztworu (i/lub zdjęcie szarego kapturka z końcówki dla postaci opakowania szpryt wstępnie napełniony) pod warunkiem przechowywania w temperaturze nie wyższej niż 25 °C.
Przechowywać w miejscu niedostępnym dla dzieci.
Niezgodność.
Ponieważ nie przeprowadzono badań zgodności tego leku, nie wolno go mieszać z innymi lekami, w tym z innymi roztworami do infuzji.
Do podania roztworu należy użyć zestawu do infuzji zawartego w opakowaniu, ponieważ czynnik krzepnięcia krwi VIII może adsorbować się na wewnętrznych powierzchniach innego sprzętu do infuzji.
Opakowanie.
Postać opakowania – fiolka:
1 fiolka z liofilizatem, 1 szpryt wstępnie napełniony z rozpuszczalnikiem, 1 adapter do fiolki, 1 zestaw do infuzji, 2 tampony alkoholowe, 1 plaster oraz 1 wacik gazowy w tekturowym pudełku.
Postać opakowania – szpryt wstępnie napełniony:
1 szpryt wstępnie napełniony z liofilizatem w komorze górnej i rozpuszczalnikiem 4 ml w komorze dolnej, 1 tłok strzykawki, 1 zestaw do infuzji, 2 tampony alkoholowe, 1 plaster, 1 wacik gazowy oraz 1 kaptur w tekturowym pudełku.
Kategoria wydawania. Na receptę.
Producent.
Wyeth Farma S.A. / Wyeth Farma S.A.
Adres producenta i miejsce prowadzenia działalności.
Autovia del Norte A1, Km 23, desvio Algete, Km. 1, San Sebastian de los Reyes, 28700 Madrid, Hiszpania / Autovia del Norte A1, Km 23, desvio Algete, Km. 1, San Sebastian de los Reyes, 28700 Madrid, Spain.