Sumamed® Forte
Ucraina
Indice
ISTRUZIONE PER L'USO MEDICINALE DEL MEDICINALE SUMAMED® FORTE (SUMAMED® FORTE)
Composizione:
sostanza attiva: azitromicina;
1 dose (5 ml) di sospensione contiene 200 mg di azitromicina sotto forma di azitromicina diidrato;
sostanze eccipienti: saccarosio, fosfato di sodio, idrossipropilcellulosa, gomma xantana, biossido di silicio colloidale anidro,
aroma(-i) e/o colorante:
aroma di banana, aroma di ciliegia, aroma di vaniglia
oppure
biossido di titanio (E 171), aroma di banana, aroma di vaniglia,
oppure
biossido di titanio (E 171), aroma di fragola,
oppure
biossido di titanio (E 171), aroma di lampone.
Forma farmaceutica. Polvere per sospensione orale.
Principali caratteristiche fisico-chimiche: polvere di colore bianco o bianco-giallastro con odore caratteristico di ciliegia e banana oppure di banana, oppure di fragola, oppure di lampone.
La sospensione ricostituita è una sospensione omogenea di colore bianco-giallastro con odore caratteristico di ciliegia e banana oppure di banana, oppure di fragola, oppure di lampone.
Gruppo farmacoterapeutico. Agenti antibatterici per uso sistemico. Macrolidi, lincosamidi e streptogrammine. Azitromicina. Codice ATC J01FA10.
Proprietà farmacologiche.
Farmacodinamica.
L'azitromicina è un antibiotico macrolidico appartenente al gruppo degli azalidi. La molecola è ottenuta inserendo un atomo di azoto nell'anello lactonico dell'eritromicina A. Il meccanismo d'azione dell'azitromicina consiste nell'inibizione della sintesi proteica batterica mediante legame alla subunità ribosomiale 50S e inibizione della traslocazione peptidica.
Meccanismo di resistenza.
Esiste una completa resistenza crociata tra Streptococcus pneumoniae, streptococco emolitico di gruppo A, Enterococcus faecalis e Staphylococcus aureus, inclusi i ceppi meticillino-resistenti (MRSA), verso eritromicina, azitromicina, altri macrolidi e lincosamidi.
La diffusione della resistenza acquisita per questi microrganismi può variare in base alla località geografica e al tempo; pertanto, è necessario disporre di informazioni locali sulla resistenza, specialmente nel trattamento delle infezioni gravi. Se necessario, si raccomanda di consultare un esperto qualificato, soprattutto quando l'entità locale della resistenza rende incerta l'efficacia del farmaco nel trattamento di almeno alcuni tipi di infezioni.
Spettro di attività antimicrobica dell'azitromicina
| Specie generalmente sensibili |
| Batteri Gram-positivi aerobi |
| Staphylococcus aureus sensibile alla meticillina |
| Streptococcus pneumoniae sensibile alla penicillina |
| Streptococcus pyogenes |
| Batteri Gram-negativi aerobi |
| Haemophilus influenzae Haemophilus parainfluenzae |
| Legionella pneumophila |
| Moraxella catarrhalis |
| Pasteurella multocida |
| Batteri anaerobi |
| Clostridium perfringens |
| Fusobacterium spp. |
| Prevotella spp. |
| Porphyriomonas spp. |
| Altri microrganismi |
| Chlamydia trachomatis Chlamydia pneumoniae Mycoplasma pneumoniae |
| Specie per cui la resistenza acquisita può rappresentare un problema |
| Batteri Gram-positivi aerobi |
| Streptococcus pneumoniae con sensibilità intermedia alla penicillina e resistente alla penicillina |
| Organismi intrinsecamente resistenti |
| Batteri Gram-positivi aerobi |
| Enterococcus faecalis |
| Stafilococchi MRSA, MRSE* |
| Batteri anaerobi |
| Gruppo di batterioidi Bacteroides fragilis |
*Metilcillina - resistente Staphylococcus aureus ha un'elevata prevalenza di resistenza acquisita ai macrolidi ed è menzionato qui per la rara sensibilità all'azitromicina.
Farmacocinetica.
La biodisponibilità dopo somministrazione orale è di circa il 37%. La concentrazione massima nel plasma sanguigno viene raggiunta entro 2-3 ore dopo l'assunzione del farmaco.
Dopo somministrazione orale, l'azitromicina si distribuisce in tutto l'organismo. Studi farmacocinetici hanno dimostrato che la concentrazione di azitromicina nei tessuti è significativamente più alta (fino a 50 volte) rispetto a quella nel plasma, indicando un forte legame del farmaco con i tessuti.
Il legame con le proteine plasmatiche varia in base alle concentrazioni plasmatiche ed è compreso tra il 12% a 0,5 µg/ml e il 52% a 0,05 µg/ml nel siero. Il volume apparente di distribuzione allo stato stazionario (VVss) è di 31,1 l/kg.
Il periodo terminale di emivita plasmatica riflette pienamente il tempo di eliminazione dai tessuti, che si protrae da 2 a 4 giorni.
Circa il 12% della dose somministrata per via endovenosa di azitromicina viene escreto immodificato nelle urine nei tre giorni successivi. Concentrazioni particolarmente elevate di azitromicina immodificata sono state riscontrate nella bile umana. Nella bile sono stati inoltre identificati dieci metaboliti, formati per demetilazione N- e O-, idrossilazione degli anelli desosamina e aglicone e scissione del coniugato cladinicosio. Il confronto dei risultati ottenuti mediante cromatografia liquida e saggi microbiologici ha dimostrato che i metaboliti dell'azitromicina non sono microbiologicamente attivi.
Caratteristiche cliniche.
Indicazioni.
Infezioni causate da microrganismi sensibili all'azitromicina:
- infezioni dell'apparato otorinolaringoiatrico (faringite/tonsillite batterica, sinusite, otite media);
- infezioni delle vie respiratorie (bronchite batterica, polmonite comunitaria);
- infezioni della cute e dei tessuti molli: eritema migrante (stadio iniziale della malattia di Lyme), escarese, impetigine, piodermiti secondarie.
Controindicazioni.
Ipersensibilità all'azitromicina, all'eritromicina, a qualsiasi antibiotico macrolidico o chetolidico, o a uno qualsiasi degli altri componenti del medicinale.
Interazioni con altri medicinali ed altre forme di interazione.
Antiacidi. Nello studio sull'effetto dell'assunzione concomitante di antiacidi sulla farmacocinetica dell'azitromicina, in generale non sono state osservate variazioni nella biodisponibilità, anche se la concentrazione plasmatica massima dell'azitromicina è diminuita di circa il 25%. Non si raccomanda l'assunzione contemporanea di azitromicina e antiacidi.
Cetirizina. In volontari sani, l'assunzione concomitante di azitromicina per 5 giorni con cetirizina 20 mg a regime di equilibrio non ha mostrato alcuna interazione farmacocinetica né variazioni significative dell'intervallo QT.
Didanosina. L'assunzione concomitante di dosi giornaliere di 1200 mg di azitromicina con 400 mg di didanosina al giorno in sei volontari sieropositivi non ha evidenziato alcun effetto sulla farmacocinetica della didanosina a regime di equilibrio rispetto al placebo.
Digossina e colchicina. È stato riportato che l'uso concomitante di antibiotici macrolidici, inclusa l'azitromicina, e substrati della glicoproteina-P, come la digossina e la colchicina, può portare ad un aumento dei livelli sierici di tali substrati. Pertanto, quando si somministra concomitante azitromicina e un substrato della glicoproteina-P come la digossina, si deve considerare la possibilità di un aumento della concentrazione sierica del substrato.
Zidovudina. Dosi singole di 1000 mg e dosi multiple di 1200 mg o 600 mg di azitromicina hanno avuto un effetto trascurabile sulla farmacocinetica plasmatica o sull'escrezione urinaria della zidovudina o dei suoi metaboliti glucuronidi. Tuttavia, l'assunzione di azitromicina ha aumentato la concentrazione di zidovudina fosforilata, il metabolita clinicamente attivo, nei monociti del sangue periferico. L'importanza clinica di questi dati non è chiara, ma potrebbe essere utile per i pazienti.
L'azitromicina non interagisce in modo significativo con il sistema epatico del citocromo P450. Si ritiene che il farmaco non presenti interazioni farmacocinetiche tipiche dell'eritromicina e di altri macrolidi. L'azitromicina non induce né inattiva il citocromo epatico P450 attraverso complessi metaboliti-citocromo.
Ergotamina. A causa della possibilità teorica di ergotismo, non è raccomandata l'assunzione concomitante di azitromicina con derivati dell'ergotamina.
Sono stati condotti studi farmacocinetici sull'uso concomitante di azitromicina e dei seguenti farmaci, il cui metabolismo avviene in larga misura tramite il citocromo P450.
Atorvastatina. L'assunzione concomitante di atorvastatina (10 mg al giorno) e azitromicina (500 mg al giorno) non ha causato variazioni nelle concentrazioni plasmatiche di atorvastatina (basate sull'analisi dell'inibizione della HMG-CoA reduttasi). Tuttavia, nel periodo post-marketing sono stati riportati casi di rabdomiolisi in pazienti che assumevano azitromicina con statine.
Carbamazepina. In uno studio di interazione farmacocinetica in volontari sani, l'azitromicina non ha mostrato un effetto significativo sui livelli plasmatici di carbamazepina o dei suoi metaboliti attivi.
Cimetidina. In uno studio farmacocinetico sull'effetto di una dose singola di cimetidina assunta 2 ore prima dell'azitromicina, non sono state osservate variazioni nella farmacocinetica dell'azitromicina.
Anticoagulanti orali di tipo cumarinico. In uno studio di interazione farmacocinetica, l'azitromicina non ha modificato l'effetto anticoagulante di una dose singola di 15 mg di warfarin somministrata a volontari sani. Nel periodo post-marketing sono stati riportati casi di potenziamento dell'effetto anticoagulante dopo l'assunzione concomitante di azitromicina e anticoagulanti orali di tipo cumarinico. Sebbene non sia stato stabilito un nesso causale, si deve considerare la necessità di un monitoraggio frequente del tempo di protrombina nei pazienti che assumono anticoagulanti orali di tipo cumarinico e ai quali viene prescritta azitromicina.
Ciclosporina. In uno studio farmacocinetico su volontari sani che hanno ricevuto una dose orale di azitromicina 500 mg/giorno per 3 giorni, seguita da una dose orale singola di ciclosporina 10 mg/kg, è stato dimostrato un significativo aumento della Cmax e dell'AUC0-5 della ciclosporina. Pertanto, si deve prestare cautela nell'uso concomitante di questi farmaci. Se l'assunzione concomitante è necessaria, si devono monitorare i livelli di ciclosporina e adeguare la dose in modo appropriato.
Efavirenz. L'assunzione concomitante di una dose singola di azitromicina 600 mg e 400 mg di efavirenz al giorno per 7 giorni non ha causato alcuna interazione farmacocinetica clinicamente significativa.
Fluconazolo. L'assunzione concomitante di una dose singola di azitromicina 1200 mg non provoca variazioni nella farmacocinetica di una dose singola di fluconazolo 800 mg. L'esposizione totale e il tempo di dimezzamento dell'azitromicina non sono modificati dall'assunzione concomitante di fluconazolo, ma è stata osservata una riduzione clinicamente non significativa della Cmax (18%) dell'azitromicina.
Indinavir. L'assunzione concomitante di una dose singola di azitromicina 1200 mg non ha avuto un effetto statisticamente significativo sulla farmacocinetica dell'indinavir, assunto alla dose di 800 mg tre volte al giorno per 5 giorni.
Metilprednisolone. In uno studio di interazione farmacocinetica su volontari sani, l'azitromicina non ha avuto un effetto significativo sulla farmacocinetica del metilprednisolone.
Midazolam. In volontari sani, l'assunzione concomitante di azitromicina 500 mg al giorno per 3 giorni non ha causato variazioni clinicamente significative nella farmacocinetica e farmacodinamica del midazolam somministrato come dose singola di 15 mg.
Nelfinavir. L'assunzione concomitante di azitromicina (1200 mg) e nelfinavir a concentrazioni di regime (750 mg tre volte al giorno) provoca un aumento della concentrazione di azitromicina. Non sono stati osservati effetti collaterali clinicamente significativi, pertanto non è necessario aggiustare la dose.
Rifabutina. L'assunzione concomitante di azitromicina e rifabutina non ha influenzato le concentrazioni di questi farmaci nel siero. È stata osservata neutropenia in soggetti che assumevano contemporaneamente azitromicina e rifabutina. Sebbene la neutropenia fosse associata all'uso di rifabutina, un nesso causale con l'assunzione concomitante di azitromicina non è stato stabilito.
Sildenafil. In volontari sani di sesso maschile, non sono state trovate evidenze che l'azitromicina (500 mg al giorno per 3 giorni) influisca sui valori di AUC e Cmax dello sildenafil o del suo principale metabolita circolante.
Terfenadina. Negli studi farmacocinetici non sono stati riportati casi di interazione tra azitromicina e terfenadina. In alcuni casi non si può escludere completamente la possibilità di tale interazione; tuttavia, non esistono dati specifici sulla sua esistenza.
Teofillina. Non sono disponibili dati su interazioni farmacocinetiche clinicamente significative tra azitromicina e teofillina in volontari sani.
Triazolam. L'assunzione concomitante in volontari sani di azitromicina 500 mg il primo giorno e 250 mg il secondo giorno con 0,125 mg di triazolam non ha avuto un effetto significativo su tutti i parametri farmacocinetici del triazolam rispetto all'assunzione di triazolam e placebo.
Trimetoprim/sulfametossazolo. L'assunzione concomitante per 7 giorni di trimetoprim/sulfametossazolo a doppia concentrazione (160 mg/800 mg) con azitromicina 1200 mg al settimo giorno non ha mostrato un effetto significativo sulle concentrazioni massime, sull'esposizione totale o sull'escrezione urinaria di trimetoprim o sulfametossazolo. I livelli sierici di azitromicina erano paragonabili a quelli osservati in altri studi.
Idrossiclorochina. L'azitromicina deve essere somministrata con cautela ai pazienti che assumono medicinali che prolungano l'intervallo QT e possono causare aritmie cardiache, come l'idrossiclorochina.
Caratteristiche nell'uso.
Come nel caso dell'eritromicina e di altri antibiotici macrolidici, sono stati segnalati rari casi di gravi reazioni allergiche, inclusi angioedema e anafilassi (in rari casi con esito fatale), reazioni dermatologiche, tra cui l'eczema esfoliativo acuto generalizzato. Alcune di queste reazioni indotte dall'azitromicina hanno causato sintomi ricorrenti e hanno richiesto un monitoraggio e un trattamento più prolungati.
Poiché il fegato rappresenta la principale via di eliminazione dell'azitromicina, il farmaco deve essere somministrato con cautela nei pazienti con gravi malattie epatiche. Sono stati segnalati casi di epatite fulminante che ha causato insufficienza epatica potenzialmente letale durante il trattamento con azitromicina. Probabilmente alcuni pazienti avevano in anamnesi patologie epatiche o avevano assunto altri farmaci epatotossici.
Devono essere effettuati esami/analisi della funzionalità epatica in caso di comparsa di segni e sintomi di disfunzione epatica, come astenia che si sviluppa rapidamente, accompagnata da ittero, urine scure, tendenza al sanguinamento o encefalopatia epatica. In caso di alterazione della funzionalità epatica, l'uso di azitromicina deve essere interrotto.
Nei pazienti che assumono derivati dell'ergotamina, la somministrazione concomitante di alcuni antibiotici macrolidici può favorire lo sviluppo rapido dell'ergotismo. Non sono disponibili dati riguardo alla possibile interazione tra derivati dell'ergotamina e azitromicina. Tuttavia, a causa della possibilità teorica di ergotismo, l'azitromicina non deve essere somministrata contemporaneamente ai derivati dell'ergotamina.
Come per l'uso di altri antibiotici, si raccomanda di monitorare la comparsa di segni di superinfezione causata da microrganismi resistenti, inclusi funghi.
Durante l'assunzione di quasi tutti gli agenti antibatterici, inclusa l'azitromicina, è stata segnalata diarrea associata a Clostridium difficile (CDAD), la cui gravità può variare da diarrea lieve a colite con esito fatale. Il trattamento con agenti antibatterici altera la flora normale del colon, favorendo la crescita eccessiva di C. difficile.
C. difficile produce le tossine A e B, che contribuiscono allo sviluppo della CDAD. I ceppi di C. difficile che iperproducono tossine sono responsabili di un aumento dell'incidenza e della mortalità, poiché queste infezioni possono risultare resistenti alla terapia antimicrobica e richiedere una colectomia. È necessario considerare la possibile insorgenza di CDAD in tutti i pazienti con diarrea indotta da antibiotici. È importante raccogliere un'accurata anamnesi, poiché la CDAD può manifestarsi fino a due mesi dopo l'assunzione di agenti antibatterici.
Nei pazienti con grave disfunzione renale (velocità di filtrazione glomerulare <10 ml/min) è stato osservato un aumento del 33% dell'esposizione sistemica all'azitromicina.
Prolungamento della ripolarizzazione cardiaca e dell'intervallo QT, che aumentano il rischio di aritmie cardiache e di fibrillazione/tachicardia ventricolare (torsade de pointes), sono stati osservati durante il trattamento con altri antibiotici macrolidici, inclusa l'azitromicina. Poiché le condizioni associate a un aumento del rischio di aritmie ventricolari (inclusa la torsade de pointes) possono portare all'arresto cardiaco, l'azitromicina deve essere somministrata con cautela ai pazienti con condizioni proaritmiche preesistenti (in particolare donne e pazienti anziani), specialmente a quei pazienti:
- con prolungamento congenito o documentato dell'intervallo QT;
- attualmente in trattamento con altre sostanze attive note per prolungare l'intervallo QT, come antiaritmici di classe IA (chinidina e procainamide) e classe III (dofetilide, amiodarone e sotalolo), cisapride e terfenadina, neurolettici come il pimozide, antidepressivi come il citalopram, e fluorochinoloni come moxifloxacina e levofloxacina;
- con alterazioni dell'equilibrio elettrolitico, in particolare in caso di ipokaliemia e ipomagnesiemia;
- con bradicardia clinicamente rilevante, aritmia cardiaca o insufficienza cardiaca grave.
Sono stati segnalati peggioramento dei sintomi di miastenia grave o insorgenza di nuovo quadro miastenico in pazienti in trattamento con azitromicina.
Nel trattamento della faringite/tonsillite causata da Streptococcus pyogenes, il farmaco di prima scelta è generalmente la penicillina, che viene utilizzata anche per la profilassi della febbre reumatica acuta. L'azitromicina è in genere efficace nel trattamento delle infezioni da streptococco nella faringe, ma non ci sono dati che dimostrino l'efficacia dell'azitromicina nella prevenzione degli episodi reumatici.
Non sono state stabilite la sicurezza e l'efficacia della somministrazione endovenosa di azitromicina per il trattamento delle infezioni nei bambini.
Non sono state stabilite la sicurezza e l'efficacia per la profilassi o il trattamento del complesso Mycobacterium avium nei bambini.
Azitromicina in polvere per sospensione orale contiene saccarosio. Il farmaco non deve essere somministrato a pazienti con rari problemi ereditari di intolleranza al fruttosio, malassorbimento da glucosio-galattosio o carenza di saccarasi-isomaltasi.
Azitromicina in polvere per sospensione orale contiene 83,7 mg/dose di fosfato di sodio. Si raccomanda cautela nell'uso nei pazienti sottoposti a dieta controllata di sodio.
Uso durante la gravidanza o l'allattamento.
Gravidanza
Non vi sono dati adeguati sull'uso di azitromicina in donne in gravidanza. Negli studi di tossicità riproduttiva negli animali non è stato osservato alcun effetto teratogeno dannoso dell'azitromicina sul feto, tuttavia il farmaco attraversa la placenta. La sicurezza dell'uso di azitromicina durante la gravidanza non è stata confermata. Pertanto, l'azitromicina deve essere somministrata durante la gravidanza solo se il beneficio supera il rischio.
Allattamento
È stato segnalato che l'azitromicina penetra nel latte materno umano, ma non sono stati condotti studi clinici adeguati e ben controllati che permettano di caratterizzare la farmacocinetica dell'escrezione dell'azitromicina nel latte materno.
Fertilità
Studi sulla fertilità condotti su ratti hanno mostrato una riduzione dell'incidenza di gravidanza dopo somministrazione di azitromicina. La rilevanza di questi dati nell'uomo è sconosciuta.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell'uso di macchinari.
Non esistono evidenze che l'azitromicina possa compromettere la capacità di guidare autoveicoli o di lavorare con macchinari; tuttavia, si deve considerare la possibilità di sviluppare effetti indesiderati come delirio, allucinazioni, capogiri, sonnolenza, perdita di coscienza o convulsioni, che potrebbero influire sulla capacità di guidare autoveicoli o di utilizzare macchinari.
Modalità e dosaggio.
Sumamed® Forte, polvere per sospensione orale, va assunto 1 volta al giorno almeno 1 ora prima o 2 ore dopo il pasto.
In caso di dimenticanza di una dose, si deve assumere il prima possibile la dose dimenticata, mentre le dosi successive devono essere assunte a intervalli di 24 ore.
Misurazione della dose
Nella confezione è incluso un siringa graduata per uso orale. Il volume della siringa è di 5 ml.
Bambini
Nelle infezioni dell'apparato ORL, delle vie respiratorie, della cute e dei tessuti molli (esclusa l'eritema migrante cronica) la dose giornaliera di azitromicina è di 10 mg/kg di peso corporeo, corrispondente a 0,25 ml/kg di peso corporeo di sospensione pronta all'uso. La durata del trattamento è di 3 giorni.
In base al peso corporeo del bambino, si raccomanda la seguente tabella di dosaggio della sospensione
Sumamed® Forte:
| Massa corporea (kg) |
Dose giornaliera di sospensione (ml) |
Frequenza di somministrazione |
Contenuto di azitromicina nella dose giornaliera di sospensione |
| 15–24 |
5 |
1 volta al giorno |
200 mg |
| 25–34 |
7,5 |
300 mg |
|
| 35–44 |
10 |
400 mg |
|
| ≥45 |
12,5 |
500 mg |
Preparazione e utilizzo della sospensione
Aggiungere al flacone contenente la polvere acqua distillata o bollita e raffreddata.
- Premere il tappo del flacone verso il basso e ruotarlo in senso antiorario.
- Con un contenitore pulito, misurare con la siringa dosatrice la quantità appropriata di acqua (9,5 o 16,5 o 20,0 ml) e aggiungerla al flacone contenente la polvere:
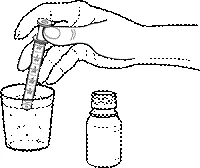
Flacone con polvere per sospensione orale 200 mg/5 ml da 15 ml: aggiungere 9,5 ml di acqua al contenuto del flacone. Agitare bene il contenuto del flacone fino ad ottenere una sospensione omogenea. Il volume della sospensione ottenuta è di circa 20 ml*.
Flacone con polvere per sospensione orale 200 mg/5 ml da 30 ml: aggiungere 16,5 ml di acqua al contenuto del flacone. Agitare bene il contenuto del flacone fino ad ottenere una sospensione omogenea. Il volume della sospensione ottenuta è di circa 35 ml*.
Flacone con polvere per sospensione orale 200 mg/5 ml da 37,5 ml: aggiungere 20 ml di acqua al contenuto del flacone. Agitare bene il contenuto del flacone fino ad ottenere una sospensione omogenea. Il volume della sospensione ottenuta è di circa 42,5 ml*.
* Dopo la dissoluzione della polvere, il flacone conterrà 5 ml aggiuntivi di sospensione (per compensare eventuali perdite durante l'uso).
| Volume del flacone |
Quantità di acqua da aggiungere al flacone per ottenere una sospensione (ml) |
| 15 ml |
9,5 |
| 30 ml |
16,5 |
| 37,5 ml |
20 |
**Le informazioni sul volume della fiala sono riportate sulla confezione e sull'etichetta della fiala.
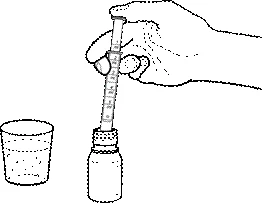
- Immergere la siringa nella sospensione e, tirando il pistone verso l'alto, misurare la quantità necessaria di sospensione.
- Se nella siringa sono presenti bolle d'aria, rimettere il medicinale nella fiala e ripetere la procedura 3.
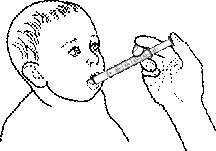
- Posizionare il bambino come per l'allattamento.
- Appoggiare la punta della siringa in bocca al bambino e somministrare lentamente il contenuto.
- Lasciare che il bambino inghiotta gradualmente tutta la quantità somministrata.
|
|
- Dopo l'assunzione del medicamento, far bere al bambino un po' di tè o succo per rimuovere e deglutire eventuali residui di sospensione nella cavità orale.
- Smontare la siringa utilizzata, lavarla con acqua corrente, asciugarla e conservarla in un luogo asciutto e pulito insieme al medicamento.
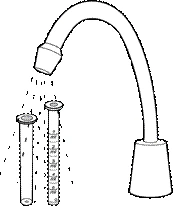
- Dopo che il bambino ha assunto l'ultima dose del medicamento, la siringa e il flacone devono essere smaltiti.
Nell’eritema migrante, la durata del trattamento è di 5 giorni. Il 1° giorno va assunta una dose di 20 mg/kg di azitromicina, corrispondente a 0,5 ml/kg di sospensione pronta all’uso. Dal 2° al 5° giorno assumere 10 mg/kg, corrispondenti a 0,25 ml/kg di sospensione pronta all’uso.
La dose totale del ciclo terapeutico è di 60 mg/kg.
| Giorni di trattamento |
1 |
2 |
3 |
4 |
5 |
| Dose giornaliera (ml/kg) |
0,5 |
0,25 |
0,25 |
0,25 |
0,25 |
È stato dimostrato che l'azitromicina è efficace nel trattamento della faringite streptococcica nei bambini, somministrata in una dose singola di 10 mg/kg o per 20 mg/kg al giorno per 3 giorni. Nelle sperimentazioni cliniche di confronto tra queste due dosi, è stata osservata una simile efficacia clinica, anche se l'eradicazione batterica è risultata maggiore con la dose giornaliera di 20 mg/kg. Tuttavia, il farmaco di scelta per la profilassi della faringite causata da Streptococcus pyogenes e per l'artrite reumatica che insorge come malattia secondaria è la penicillina.
Adulti
Nelle infezioni dell'apparato otomatolaringoiatrico e delle vie respiratorie, della cute e dei tessuti molli (esclusa l'eritema migrante cronico) la dose totale di azitromicina è di 1500 mg: 500 mg una volta al giorno. La durata del trattamento è di 3 giorni.
Nell'eritema migrante la dose totale di azitromicina è di 3 g: 1 g nel primo giorno, seguiti da 500 mg una volta al giorno dal secondo al quinto giorno. La durata del trattamento è di 5 giorni.
Pazienti anziani
Nei pazienti anziani non è necessario modificare il dosaggio.
Poiché i pazienti anziani possono appartenere a gruppi a rischio per alterazioni della conduzione cardiaca, si raccomanda cautela nell'uso dell'azitromicina a causa del rischio di sviluppare aritmie cardiache e aritmia tipo torsade de pointes.
Pazienti con compromissione renale
Nei pazienti con lieve compromissione renale (velocità di filtrazione glomerulare 10–80 ml/min) si può utilizzare lo stesso dosaggio previsto per pazienti con funzionalità renale normale. L'azitromicina deve essere somministrata con cautela nei pazienti con grave compromissione renale (velocità di filtrazione glomerulare < 10 ml/min).
Pazienti con compromissione epatica
Poiché l'azitromicina è metabolizzata nel fegato ed escreta con la bile, il farmaco non deve essere somministrato ai pazienti con grave compromissione epatica. Non sono stati condotti studi clinici sul trattamento di tali pazienti con azitromicina.
Pediatria.
Nei bambini con peso corporeo inferiore a 15 kg si raccomanda la somministrazione di Sumamed® (100 mg/5 ml). Sumamed® Forte è indicato nei bambini con peso corporeo superiore a 15 kg.
Sovradosaggio.
L'esperienza clinica con l'azitromicina indica che gli effetti indesiderati che si manifestano in seguito all'assunzione di dosi superiori a quelle raccomandate sono simili a quelli osservati con le normali dosi terapeutiche. Tali effetti possono includere diarrea, nausea, vomito e perdita reversibile dell'udito. In caso di sovradosaggio, se necessario, si raccomanda l'assunzione di carbone attivo e l'adozione di misure terapeutiche sintomatiche e di supporto generali.
Effetti indesiderati
Nella tabella sottostante, gli effetti indesiderati osservati con tutte le forme farmaceutiche di azitromicina sono elencati in base alle classi di sistema e organo e alla frequenza di insorgenza, sulla base dei dati provenienti dagli studi clinici e dal periodo di sorveglianza post-marketing, osservati con l'uso dell'azitromicina. Gli effetti indesiderati segnalati durante il periodo di sorveglianza post-marketing sono riportati in corsivo. Le categorie di frequenza sono definite secondo la seguente scala: molto comune (≥ 1/10); comune (≥ 1/100 a < 1/10); non comune (≥ 1/1.000 a < 1/100); raro (≥ 1/10.000 a < 1/1.000); molto raro (< 1/10.000); non noto (non può essere determinato dai dati disponibili). Entro ciascuna categoria di frequenza, gli eventi avversi sono elencati in ordine decrescente di gravità.
Effetti indesiderati possibili o probabili correlati all'azitromicina, sulla base dei dati ottenuti durante gli studi clinici e nel periodo di sorveglianza post-marketing
| Classe di sistema e organo |
Reazione avversa |
Frequenza |
| Infezioni e infestazioni |
Candidosi, infezioni vaginali, polmonite, infezione fungina, infezione batterica, faringite, gastroenterite, disturbi della funzione respiratoria, rinite, candidosi orale |
Non comune |
| Colite pseudomembranosa |
Sconosciuta |
|
| Sistema emolinfopoietico |
Leucopenia, neutropenia, eosinofilia |
Non comune |
| Trombocitopenia, anemia emolitica |
Sconosciuta |
|
| Sistema immunitario |
Edema angioneurotico, reazioni di ipersensibilità |
Non comune |
| Reazione anafilattica |
Sconosciuta |
|
| Disturbi del metabolismo e della nutrizione |
Anoressia |
Non comune |
| Disturbi psichici |
Nervosismo, insonnia |
Non comune |
| Agitazione |
Raro |
|
| Aggressività, ansia, delirio, allucinazioni |
Sconosciuta |
|
| Sistema nervoso |
Cefalea |
Comune |
| Vertigini, sonnolenza, disgeusia, parastesia |
Non comune |
|
| Perdita di coscienza, convulsioni, ipoestesia, attività psicomotoria aumentata, anosmia, ageusia, parosmia, miastenia grave |
Sconosciuta |
|
| Organi della vista |
Disturbi della vista |
Non comune |
| Organi dell'udito |
Disturbi dell'udito, vertigini |
Non comune |
| Disturbi dell'udito, inclusa sordità e/o acufene |
Sconosciuta |
|
| Apparato cardiaco |
Palpitazioni |
Non comune |
| Arritmia ventricolare (torsione di punta), aritmia, inclusa tachicardia ventricolare, prolungamento dell'intervallo QT nell'ECG |
Sconosciuta |
|
| Vasi sanguigni |
Flush |
Non comune |
| Ipotensione arteriosa |
Sconosciuta |
|
| Apparato respiratorio |
Dispnea, epistassi |
Non comune |
| Apparato gastrointestinale |
Diarrea |
Molto comune |
| Vomito, dolore addominale, nausea |
Comune |
|
| Stipsi, meteorismo, dispepsia, gastrite, disfagia, distensione addominale, bocca secca, eruttazione, ulcere orali, ipersalivazione |
Non comune |
|
| Pancreatite, cambiamento del colore della lingua |
Sconosciuta |
|
| Sistema epatobiliare |
Alterazione della funzionalità epatica, ittero colestatico |
Raro |
| Insufficienza epatica (raramente con esito fatale), epatite fulminante, necrosi epatica |
Sconosciuta |
|
| Pelle e tessuto sottocutaneo |
Eruzione cutanea, prurito, orticaria, dermatite, secchezza cutanea, iperidrosi |
Non comune |
| Fotosensibilità, pustolosi esantematica acuta generalizzata |
Raro |
|
| Sindrome di Stevens-Johnson, necrolisi epidermica tossica, eritema multiforme, reazione da farmaco con eosinofilia e sintomi sistemici |
Sconosciuta |
|
| Apparato muscoloscheletrico |
Osteoartrite, mialgia, dolore alla schiena, dolore al collo |
Non comune |
| Artralgia |
Sconosciuta |
|
| Apparato urinario |
Disuria, dolore renale |
Non comune |
| Insufficienza renale acuta, nefrite interstiziale |
Sconosciuta |
|
| Apparato riproduttivo e ghiandole mammarie |
Emorragia uterina, disturbi testicolari |
Non comune |
| Disturbi generali e condizioni locali |
Edema, astenia, malessere, affaticamento, edema facciale, dolore toracico, ipertermia, dolore, edema periferico |
Non comune |
| Esami di laboratorio |
Diminuzione del numero di linfociti, aumento del numero di eosinofili, diminuzione del livello di bicarbonato nel sangue, aumento del livello di basofili, aumento del livello di monociti, aumento del livello di neutrofili |
Comune |
| Aumento del livello di aspartato aminotransferasi, alanina aminotransferasi, bilirubina nel sangue, urea nel sangue, creatinina nel sangue; alterazioni dei livelli di potassio nel sangue, aumento del livello di fosfatasi alcalina, cloruro, glucosio, piastrine; diminuzione del livello di ematocrito; aumento del livello di bicarbonato, alterazione del livello di sodio |
Non comune |
|
| Lesioni e avvelenamenti |
Complicazioni post-procedura |
Non comune |
Le informazioni sulle reazioni avverse, possibilmente correlate alla profilassi e al trattamento del MycobacteriumAviumComplex, si basano su dati di studi clinici e osservazioni nel periodo post-marketing. Queste reazioni avverse differiscono per tipo o frequenza rispetto a quelle riportate con l'uso di forme farmaceutiche a rilascio immediato e a rilascio prolungato.
Reazioni avverse, possibilmente correlate alla profilassi e al trattamento del MycobacteriumAviumComplex
| Classe di sistema e di organo |
Reazione avversa |
Frequenza |
||
| Disturbi del metabolismo |
Anoressia |
Comune |
||
| Disturbi del sistema nervoso |
Capogiri, cefalea, parastesia, disgeusia |
Comune |
||
| Ipestesia |
Non comune |
|||
| Disturbi della vista |
Disturbi visivi |
Comune |
||
| Disturbi dell'udito |
Sordità |
Comune |
||
| Disturbi dell'udito, acufene |
Non comune |
|||
| Disturbi cardiaci |
Palpitazioni |
Non comune |
||
| Disturbi gastrointestinali |
Diarea, dolore addominale, nausea, meteorismo, fastidio gastrointestinale, evacuazioni frequenti e liquide |
Molto comune |
||
| Disturbi del sistema epatobiliare |
Epatite |
Non comune |
||
| Disturbi della pelle e del tessuto sottocutaneo |
Eruzione cutanea, prurito |
Comune |
||
| Sindrome di Stevens-Johnson, fotosensibilità |
Non comune |
|||
| Disturbi del sistema muscoloscheletrico |
Artralgia |
Comune |
||
| Disturbi sistemici e reazioni locali |
Aumentata affaticabilità |
Comune |
||
| Astenia, malessere |
Non comune |
|||
Durata della validità. 2 anni.
Durata della validità della sospensione pronta all'uso – 5 giorni (15 ml), 10 giorni (30 ml, 37,5 ml).
Condizioni di conservazione.
Conservare a temperatura non superiore a 25 °C, in un luogo inaccessibile ai bambini.
Conservare la sospensione pronta all'uso a temperatura non superiore a 25 °C.
Confezione.
1 flacone con polvere per sospensione orale da 15 ml (600 mg) oppure da 30 ml (1200 mg) oppure da 37,5 ml (1500 mg), insieme ad una siringa dosatrice, in una confezione.
Categoria di vendita. Sotto prescrizione medica.
Produttore. PLIVA Hrvatska d.o.o.
Sede del produttore ed indirizzo del luogo dell'attività commerciale.
Prijelaz baruna Filipovića 25, 10000 Zagabria, Croazia.