Somavert
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE Somavert (Somavert)
Composizione:
Principio attivo: pegvisomant;
1 flaconcino contiene 10 mg, 15 mg, 20 mg o 30 mg di pegvisomant;
Eccipienti: glicina, mannitolo (E 421), fosfato monosodico anidro, fosfato disodico monoidrato.
1 siringa preriempita con solvente contiene acqua per preparazioni iniettabili.
Forma farmaceutica. Liofilizzato e solvente per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: massa liofilizzata da bianca a quasi bianca.
Categoria farmacoterapeutica. Altri ormoni dell'ipofisi anteriore e loro analoghi. Pegvisomant. Codice ATC H01A X01.
Proprietà farmacologiche
Descrizione
Somavert contiene pegvisomant, un analogo dell'ormone della crescita umano che è stato modificato strutturalmente per agire come antagonista dei recettori dell'ormone della crescita.
Il pegvisomant è una proteina prodotta mediante tecnologia del DNA ricombinante, costituita da un residuo di 191 aminoacidi al quale sono legati covalentemente diversi polimeri di polietilenglicole (PEG) (principalmente da 4 a 6 molecole di PEG/proteina). La massa molecolare della proteina pegvisomant è di 21.998 Dalton. La massa molecolare della parte PEG del pegvisomant è di circa 5.000 Dalton. Di conseguenza, la massa molecolare del pegvisomant è prevalentemente di circa 42.000; 47.000 e 52.000 Dalton. Il pegvisomant è prodotto da un ceppo specifico di batteri Escherichia coli geneticamente modificati mediante l'inserimento di un plasmide contenente il gene per l'antagonista del recettore dell'ormone della crescita. L'attività biologica è determinata mediante un saggio biologico di proliferazione cellulare. Il legame del farmaco Somavert con il recettore dell'ormone della crescita determina l'alterazione del normale legame con un secondo recettore dell'ormone della crescita, inibendo la dimerizzazione del recettore funzionale e il successivo percorso di segnalazione intracellulare.
Farmacodinamica
Il pegvisomant si lega in modo selettivo al recettore dell'ormone della crescita sulla superficie cellulare e non presenta reattività crociata con altri 19 recettori citochinici studiati, inclusa la prolattina. Il pegvisomant determina una riduzione della concentrazione plasmatica del fattore di crescita simile all'insulina I (IGF-1), dell'IGF-1 libero, della subunità acido-labile (ALS) e della proteina di legame del fattore di crescita simile all'insulina di tipo 3 (IGFBP-3) (vedi sotto «Studi clinici», Fig. 1).
Meccanismo d'azione
Il pegvisomant si lega in modo selettivo ai recettori dell'ormone della crescita sulla superficie cellulare, bloccando il legame dell'ormone della crescita endogeno e impedendo così la trasmissione del segnale dell'ormone della crescita.
L'inibizione dell'azione dell'ormone della crescita determina una riduzione della concentrazione plasmatica dell'IGF-1, nonché di altre proteine plasmatiche sensibili all'ormone della crescita, come l'IGF-1 libero, la subunità acido-labile dell'IGF-1 e l'IGFBP-3.
Studi clinici
Complessivamente, 112 pazienti (63 uomini e 49 donne) con acromegalia hanno partecipato a uno studio multicentrico, randomizzato, in doppio cieco, della durata di 12 settimane, volto a confrontare placebo e Somavert. L'età media ± deviazione standard (DS) era di 48 ± 14 anni e la durata media dell'acromegalia era di 8 ± 8 anni. A 93 pazienti era stata precedentemente eseguita un'intervento chirurgico all'ipofisi, di cui a 57 era stata inoltre somministrata una radioterapia standard. A 6 pazienti era stata somministrata radioterapia senza intervento chirurgico, a 9 era stata somministrata solo terapia farmacologica e a ulteriori 4 pazienti non era stata somministrata alcuna terapia in precedenza. Al momento dell'inizio dello studio, il tempo medio ± DS trascorso dall'ultima chirurgia e/o radioterapia era rispettivamente di 6,8 ± 0,93 anni (n = 63) e 5,6 ± 0,57 anni (n = 93).
I pazienti sono stati inclusi nello studio se presentavano un livello di IGF-1 nel siero, determinato dopo un adeguato periodo di washout del farmaco, superiore di almeno 1,3 volte il limite superiore della norma, corretto per età. Al momento della visita di inclusione, i pazienti sono stati assegnati in modo randomizzato a uno dei quattro gruppi terapeutici: placebo (n = 32); Somavert per via sottocutanea alla dose di 10 mg/giorno (n = 26), 15 mg/giorno (n = 26) o 20 mg/giorno (n = 28). Il parametro primario di efficacia era la variazione percentuale della concentrazione di IGF-1 dal livello alla visita di inclusione fino alla settimana 12. Nei tre gruppi trattati con Somavert si è osservata una riduzione statisticamente significativa (p < 0,01) del livello di IGF-1 nel siero rispetto al gruppo placebo (Tab. 1).
Tabella 1. Variazione percentuale media della concentrazione di IGF-1 dal livello alla visita di inclusione fino alla settimana 12 nella popolazione «pazienti trattati»
| Indici |
Placebo n = 31 |
Somavert |
||
| 10 mg/giorno n = 26 |
15 mg/giorno n = 26 |
20 mg/giorno n = 28 |
||
| Concentrazione media di IGF-1 al momento della visita di inclusione (ng/ml) (SV) |
670 (288) |
627 (251) |
649 (293) |
732 (205) |
| Variazione percentuale media della concentrazione di IGF-1 rispetto alla visita di inclusione (SV) |
|
|
|
|
| Somavert meno placebo (95 % IC per la differenza tra i farmaci in studio) |
(–35; –11) |
(–56; –33) |
(–68; –49) |
|
* P < 0,01; n – numero di pazienti; DS – deviazione standard.
Inoltre, nei gruppi trattati con Somavert si è osservata una riduzione dei livelli sierici di IGF-1 libero, IGFBP-3 e BAP rispetto al gruppo placebo in tutti i momenti delle visite pianificate dello studio (fig. 1).
| Somavert 15 mg/giorno (n = 24–26) Somavert 20 mg/giorno (n = 27–28) |
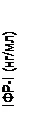
| 800 600 400 200 |
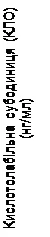
| Placebo (n = 31) Somavert 10 mg/giorno (n = 25–26) |
| 0 2 4 8 12 Settimana |
| 0 2 4 8 12 Settimana |
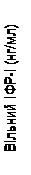
Fig. 1. Effetto del medicinale Somavert sui marcatori nel siero (media ± errore standard della media).
Dopo 12 settimane di terapia, sono state osservate concentrazioni normalizzate di IGF-1 nel seguente numero di pazienti (in percentuale) (fig. 2).
| Somavert 20 mg/giorno |
| Somavert 15 mg/giorno |
| Somavert 10 mg/giorno |
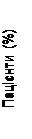
| Placebo |
Fig. 2. Percentuale di pazienti nei quali i livelli di IGF-1 sono tornati alla normalità alla settimana 12 di trattamento.
In Tab. 2 è riportato l'effetto della terapia con il medicinale Somavert in termini di dimensione dell'anello (taglie standard di gioielli convertite in una scala numerica da 1 a 63 punti), nonché in base ai segni e sintomi dell'acromegalia. Ciascun singolo segno o sintomo dell'acromegalia (edema dei tessuti molli, artralgia, cefalea, sudorazione e affaticamento) è stato valutato mediante una scala ordinale a nove punti (0 = assente, 8 = grave e invalidante); il punteggio totale per la valutazione dei segni e sintomi dell'acromegalia è stato ottenuto sommando i punteggi delle singole scale. I punteggi medi al basale erano i seguenti: dimensione dell'anello – 47,1; punteggio totale per i segni e sintomi – 15,2; edema dei tessuti molli – 2,5; artralgia – 3,2; cefalea – 2,4; sudorazione – 3,3 e affaticamento – 3,7.
Tabella 2. Variazione media della dimensione dell'anello e dei segni e sintomi dell'acromegalia alla settimana 12 rispetto alla visita di inclusione nello studio (baseline).
| Indici |
Placebo n = 30 |
Somavert |
||
| 10 mg/giorno n = 26 |
15 mg/giorno n = 24–25 |
20 mg/giorno n = 26–27 |
||
| Dimensione dell'anello |
|
|
|
|
| Punteggio totale nella valutazione dei segni e sintomi dell'acromegalia |
1,3 (6,0) |
|
|
|
| Edema dei tessuti molli |
0,3 (2,3) |
|
|
|
| Artralgia |
0,1 (1,8) |
|
|
|
| Cefalea |
0,1 (1,7) |
|
|
|
| Traspirazione eccessiva |
0,1 (1,7) |
|
|
|
| Affaticamento aumentato |
0,7 (1,5) |
|
|
|
Le concentrazioni sieriche dell'ormone della crescita, misurate mediante metodi quantitativi sviluppati per lo studio e che utilizzano anticorpi non reattivi in modo incrociato con pegvisomant, aumentavano entro due settimane dall'inizio della terapia con Somavert. L'aumento maggiore delle concentrazioni di ormone della crescita si osservava nei pazienti trattati con Somavert alla dose di 20 mg/giorno. È molto probabile che questo effetto sia dovuto alla riduzione dell'inibizione della secrezione dell'ormone della crescita in seguito alla diminuzione dei livelli di IGF-1. Come mostrato in Fig. 3, quando i pazienti con acromegalia assumevano una dose iniziale di carico di Somavert seguita da dosi giornaliere fisse, l'aumento della concentrazione di ormone della crescita era inversamente proporzionale alla riduzione dei livelli di IGF-1 e si stabilizzava generalmente entro la settimana 2. La concentrazione sierica dell'ormone della crescita rimaneva stabile nei pazienti trattati con Somavert per una media di 43 settimane (intervallo 0-82 settimane).
| Placebo |
| Sett. Sett. Sett. Sett. Sett. 0 2 4 8 12 |
| 20 mg/g |
| 15 mg/g |
| 10 mg/g |

Fig. 3. Variazione percentuale delle concentrazioni di ormone della crescita e di IGF-1 nel siero.
Alla fase aperta e prolungata dello studio clinico hanno partecipato 109 pazienti (inclusi 6 nuovi pazienti), con una durata media di trattamento di 42,6 settimane (intervallo da 1 giorno a 82 settimane); tra questi, reazioni avverse si sono verificate in 93 (85,3 %) pazienti; in 16 (14,7 %) sono state osservate reazioni avverse gravi (RAG), e in 4 (3,7 %) il trattamento è stato interrotto a causa di reazioni avverse (cefalea, aumento dei livelli di parametri biochimici della funzionalità epatica, cancro del pancreas e aumento di peso). Nel complesso, 100 (92,6 %) dei 108 partecipanti con dati disponibili sulle misurazioni dell'IGF-1 hanno mostrato concentrazioni normali di IGF-1 in almeno una visita durante lo studio.
In diversi studi, nonché nello studio ACROSTUDY, il pegvisomant ha normalizzato i livelli di IGF-1 in una grande percentuale di pazienti (> 70 %) e ha ridotto significativamente i livelli di glucosio plasmatico a digiuno (GPD) e di insulina plasmatica a digiuno (IPD).
Il pegvisomant migliora anche la sensibilità all'insulina, probabilmente a causa del blocco dei recettori dell'ormone della crescita nei tessuti, principalmente nel fegato, ma anche nel tessuto adiposo, nei reni e nei muscoli scheletrici, eliminando così l'effetto dannoso dell'ormone della crescita sulle vie di segnalazione dell'insulina, sulla lipolisi e sulla gluconeogenesi. Tuttavia, il meccanismo d'azione di questo effetto non è noto con certezza. Nei pazienti diabetici con acromegalia potrebbe essere necessario ridurre le dosi di insulina o di farmaci ipoglicemizzanti (vedere sezioni «\u003cb\u003eSpeciali avvertenze e precauzioni di impiego\u003c/b\u003e» e «\u003cb\u003eInterazioni con altri medicinali ed altre forme di interazione\u003c/b\u003e»).
Farmacocinetica.
Assorbimento. Dopo somministrazione sottocutanea, la concentrazione massima di pegvisomant nel siero viene generalmente raggiunta non prima di 33–77 ore dopo l'applicazione. Il grado medio di assorbimento della dose sottocutanea di 20 mg è del 57 % rispetto a quello dopo somministrazione endovenosa della dose di 10 mg.
Distribuzione. Il volume apparente medio di distribuzione del pegvisomant è di 7 l (coefficiente di variazione 12 %), indicando una limitata distribuzione nei tessuti. Dopo somministrazione sottocutanea singola, l'esposizione (Cmax, AUC) al pegvisomant aumenta in modo non proporzionale all'aumento della dose. I valori medi ± errore standard della media (ESM) della concentrazione di pegvisomant nel siero dopo 12 settimane di somministrazione giornaliera di dosi di 10, 15 e 20 mg sono stati rispettivamente di 6600 ± 1330; 16 000 ± 2200 e 27 000 ± 3100 ng/ml.
La biodisponibilità relativa della dose di 1 × 30 mg di pegvisomant è stata confrontata con quella della dose di 2 × 15 mg di pegvisomant in uno studio con somministrazione singola. I valori di AUCinf e Cmax del pegvisomant dopo somministrazione come singola iniezione da 30 mg sono risultati approssimativamente del 6 % e del 4 % più elevati rispettivamente rispetto a quelli dopo somministrazione come due iniezioni da 15 mg ciascuna.
Metabolismo ed eliminazione. La molecola di pegvisomant contiene polimeri di polietilenglicole legati covalentemente, al fine di ridurre la velocità di clearance. La clearance del pegvisomant dopo somministrazione ripetuta è inferiore rispetto a quella osservata dopo somministrazione singola. La clearance sistemica totale media del pegvisomant nell'organismo dopo somministrazione ripetuta è stimata nell'intervallo da 36 a 28 ml/ora per dosi sottocutanee nell'intervallo da 10 a 20 mg al giorno rispettivamente. È stato osservato che la clearance del pegvisomant aumenta in relazione alla massa corporea. Il pegvisomant viene eliminato dal siero con un'emivita media compresa tra 60 e 138 ore dopo somministrazione singola o ripetuta. Meno dell'1 % della dose somministrata viene ritrovato nell'urina entro 96 ore. I percorsi di eliminazione del pegvisomant nell'uomo non sono stati studiati.
Studi di interazione tra medicinali
Negli studi clinici, ai pazienti che assumevano oppiacei era spesso necessaria una concentrazione ematica più elevata di pegvisomant per ottenere un adeguato inibizione dell'IGF-1 rispetto ai pazienti che non assumevano oppiacei. Il meccanismo di questa interazione non è noto (vedere sezione «\u003cb\u003eInterazioni con altri medicinali ed altre forme di interazione\u003c/b\u003e»).
Gruppi specifici di pazienti
Non sono stati condotti studi farmacocinetici su pazienti con compromissione della funzionalità renale, con compromissione della funzionalità epatica, su pazienti anziani o su pazienti pediatrici. L'effetto dell'appartenenza razziale sulla farmacocinetica del pegvisomant non è stato studiato. Nell'ambito di un'analisi farmacocinetica di popolazione, non sono state osservate differenze nella farmacocinetica del pegvisomant in base al sesso dei pazienti.
\u003cu\u003eImmunogenicità\u003c/u\u003e
La frequenza osservata di anticorpi contro il farmaco dipende in larga misura dalla sensibilità e specificità del metodo analitico. Le differenze nei metodi analitici escludono un confronto significativo della frequenza di anticorpi negli studi descritti di seguito con la frequenza di anticorpi in altri studi, inclusi quelli sul medicinale Somavert o altri analoghi dell'ormone della crescita.
Negli studi clinici pre-registrazione, circa il 17 % dei pazienti trattati con Somavert ha sviluppato bassi titoli di anticorpi neutralizzanti contro l'ormone della crescita. Sebbene la presenza di questi anticorpi sembri non influire sull'efficacia di Somavert, il significato clinico dello sviluppo di questi anticorpi a lungo termine non è noto. Attualmente non sono disponibili kit diagnostici per l'analisi degli anticorpi contro il pegvisomant nei pazienti in trattamento con Somavert.
I dati riportati sopra riflettono la percentuale di pazienti nei quali i risultati degli esami sono risultati positivi per anticorpi contro il medicinale Somavert. La rilevazione della formazione di anticorpi dipende in larga misura dalla sensibilità e specificità del metodo analitico. Inoltre, la frequenza osservata di risultati positivi agli esami per anticorpi potrebbe essere influenzata da diversi fattori, tra cui la manipolazione dei campioni, il momento del prelievo, i farmaci concomitanti e la patologia di base. Per questo motivo, il confronto della frequenza di formazione di anticorpi contro Somavert con quella di altri farmaci potrebbe portare a risultati fuorvianti.
Caratteristiche cliniche.
Indicazioni.
Somavert è indicato per il trattamento dell'acromegalia in pazienti che non hanno risposto adeguatamente all'intervento chirurgico o alla radioterapia, o per i quali tali terapie non sono appropriate. L'obiettivo del trattamento è la normalizzazione dei livelli sierici del fattore di crescita simile all'insulina 1 (IGF-1).
Controindicazioni.
Non deve essere usato il medicinale Somavert in caso di allergia alla sostanza attiva o a qualsiasi altro componente del medicinale.
Interazioni con altri medicinali ed altre forme di interazione.
Insulina e/o ipoglicemizzanti orali
Nei pazienti con acromegalia e diabete mellito che assumono insulina e/o ipoglicemizzanti orali, potrebbe essere necessario ridurre la dose di insulina e/o di ipoglicemizzanti orali dopo l'inizio della terapia con Somavert (vedere il paragrafo «Informazioni importanti sull’uso del medicinale»).
Oppiacei
Negli studi clinici, ai pazienti che assumevano oppiacei è spesso stata richiesta una dose più elevata di Somavert per normalizzare la concentrazione di IGF-1, rispetto ai pazienti che non assumevano oppiacei. Il meccanismo di questa interazione non è noto.
Caratteristiche di impiego.
Ipotensione associata alla riduzione dell'ormone della crescita in pazienti con diabete mellito
L'ormone della crescita contrasta l'effetto dell'insulina sul metabolismo dei carboidrati riducendo la sensibilità all'insulina. Pertanto, in alcuni pazienti trattati con Somavert, può migliorare la tolleranza al glucosio. È necessario monitorare attentamente i pazienti con diabete mellito e, se necessario, ridurre la dose dei farmaci antidiabetici per evitare episodi di ipoglicemia.
Epatotossicità
Prima di iniziare il trattamento con Somavert, è necessario valutare i livelli basali di alanina aminotransferasi (ALT), aspartato aminotransferasi (AST), bilirubina totale e fosfatasi alcalina nel siero. Nella Tabella 3 sono riportate le raccomandazioni per l'inizio del trattamento con Somavert in base ai risultati della valutazione dei parametri di funzionalità epatica (PFE).
Un aumento transitorio e asintomatico dei livelli delle transaminasi fino a 15 volte superiore al limite superiore della norma (LSN) è stato osservato in meno del 2% dei pazienti durante due studi aperti (coinvolgenti complessivamente 147 pazienti). Questi aumenti elevati delle transaminasi non sono stati associati ad un incremento della bilirubina. I livelli elevati di transaminasi sono tornati alla normalità nel tempo, spesso dopo una temporanea interruzione del trattamento. Le segnalazioni post-commercializzazione hanno evidenziato un aumento delle transaminasi epatiche nel siero superiore a 20 volte il LSN, associato ad un aumento della bilirubina totale superiore a due volte il LSN. In molti di questi casi, l'interruzione del trattamento con Somavert ha portato alla riduzione o alla normalizzazione dei parametri epatici alterati.
Durante il trattamento, Somavert deve essere utilizzato in conformità con le informazioni relative alle alterazioni dei parametri biochimici di funzionalità epatica riportate nella Tabella 4.
Tabella 3. Raccomandazioni per l'inizio del trattamento con Somavert in base ai parametri biochimici di funzionalità epatica (PFE) basali e per il monitoraggio periodico dei PFE durante il trattamento con Somavert
| Livello basale di AFP |
Raccomandazioni |
| Normale |
|
| Aumentato, ma minore o uguale a 3 volte il LSN |
È possibile utilizzare Somavert, ma è necessario monitorare l'AFP mensilmente per almeno un anno dopo l'inizio del trattamento, quindi 2 volte all'anno nell'anno successivo. |
| Superiore a 3 volte il LSN |
|
Se durante il trattamento con Somavert il paziente presenta un aumento dei livelli di FGP o qualsiasi altro segno o sintomo di alterazione della funzionalità epatica, si raccomanda il trattamento indicato di seguito (tab. 4).
Tabella 4. Raccomandazioni cliniche basate sui risultati della valutazione dei parametri biochimici della funzionalità epatica durante il trattamento con Somavert
| Livello di FGP e segni/sintomi clinici |
Raccomandazioni |
| Uguale o superiore a 3 volte il LSN, ma inferiore a 5 volte il LSN (senza segni/sintomi di epatite o altro danno epatico o senza aumento della bilirubina totale nel siero) |
|
| Almeno 5 volte superiore al LSN oppure aumento delle transaminasi di almeno 3 volte rispetto al LSN, associato a qualsiasi aumento della bilirubina totale nel siero (con o senza segni/sintomi di epatite o altro danno epatico) |
|
| Segni o sintomi che indicano epatite o altro danno epatico (ad esempio ittero, bilirubinuria, affaticamento marcato, nausea, vomito, dolore nell’ipocondrio destro, ascite, edema di origine sconosciuta, tendenza alla formazione di ematomi) |
|
I pazienti devono effettuare esami del sangue per valutare il livello di IGF-1 e i parametri biochimici della funzionalità epatica prima e durante il trattamento con Somavert; la dose del farmaco può essere modificata in base ai risultati di questi esami.
Reattività incrociata con i test quantitativi dell'ormone della crescita
Somavert presenta una struttura simile all'ormone della crescita, pertanto può dare reazioni incrociate nei test quantitativi per l'ormone della crescita. Poiché la concentrazione di Somavert nel siero, alle dosi terapeuticamente efficaci, è generalmente da 100 a 1000 volte superiore alla concentrazione reale dell'ormone della crescita nel siero dei pazienti con acromegalia, la misurazione della concentrazione di ormone della crescita nel siero può produrre valori falsamente elevati.
Crescita dei tumori produttori di ormone della crescita
Poiché i tumori ipofisari produttori di ormone della crescita possono talvolta espandersi, causando complicazioni serie (ad esempio deficit del campo visivo), è fondamentale che tutti i pazienti vengano attentamente monitorati. In caso di segni di crescita tumorale, potrebbero essere indicate procedure alternative.
Lipohipertrofia
Sono stati osservati casi di lipohipertrofia in pazienti trattati con Somavert. In uno studio in doppio cieco, controllato con placebo, della durata di 12 settimane, è stato registrato un caso (1,3%) di lipohipertrofia nel sito di iniezione in un paziente che riceveva 10 mg/giorno. Il fenomeno è scomparso continuando la terapia. In due studi aperti (coinvolgenti complessivamente 147 pazienti) si sono verificati due casi di lipohipertrofia (entrambi nei pazienti che ricevevano 10 mg/giorno). In un caso il fenomeno è regredito durante il trattamento, nell'altro ha portato all'interruzione della terapia. Per prevenire la lipohipertrofia, è necessario cambiare quotidianamente il sito di iniezione (cioè iniettare il farmaco in un punto diverso rispetto a quello utilizzato precedentemente).
Compromissione renale
Somavert non è stato studiato nei pazienti con compromissione renale e la sicurezza ed efficacia in questi pazienti non sono note.
Uso nei pazienti di età avanzata
Il numero di partecipanti di età pari o superiore a 65 anni negli studi clinici con Somavert non è stato sufficiente per determinare se la risposta di questi pazienti differisca da quella dei pazienti più giovani. In generale, la dose nei pazienti anziani deve essere scelta con cautela, iniziando solitamente dal limite inferiore dell'intervallo di dosaggio, poiché in questa fascia di età è più frequente un ridotto funzionamento del fegato, dei reni o del cuore, e sono più comuni le malattie concomitanti o l'assunzione di altri farmaci.
Contenuto di sodio
Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose. Ai pazienti che seguono una dieta iposodica si può comunicare che questo medicinale può considerarsi privo di sodio.
Uso durante la gravidanza o l'allattamento
Gravidanza
Valutazione del rischio
I dati post-marketing sull'uso di Somavert in donne in gravidanza non sono sufficienti per stabilire un legame con il rischio di malformazioni congenite gravi, aborto spontaneo o esiti avversi per la madre e il feto legati all'uso del farmaco. Il decorso clinico dell'acromegalia può migliorare durante la gravidanza (vedere la sezione «Dati clinici»). Negli studi di riproduzione sugli animali, è stata osservata fetotossicità alla dose di 6 volte superiore alla dose massima raccomandata nell'uomo, calcolata in base alla superficie corporea, dopo somministrazione sottocutanea di pegvisomant durante l'organogenesi o nel periodo precedente l'impianto (vedere la sezione «Dati»). Il rischio di fondo stimato di malformazioni congenite gravi e di aborto spontaneo nella popolazione bersaglio non è noto. Nella popolazione generale statunitense, il rischio di fondo stimato di malformazioni congenite gravi e di aborto spontaneo nelle gravidanze clinicamente riconosciute è rispettivamente del 2-4% e del 15-20%.
Dati clinici
Rischio materno e/o embrio-fetale legato alla malattia
Dati pubblicati da singoli casi, serie di casi e da uno studio interventistico di piccole dimensioni su donne in gravidanza con acromegalia hanno mostrato che il decorso clinico dell'acromegalia può migliorare o stabilizzarsi senza trattamento durante la gravidanza, specialmente se l'acromegalia è stata trattata prima della gravidanza stessa. In rari casi, l'acromegalia può peggiorare durante la gravidanza. Poiché i livelli ormonali possono variare fisiologicamente durante la gravidanza e l'interpretazione dei livelli di ormone della crescita nelle donne in gravidanza con acromegalia può essere inaffidabile, si raccomanda un monitoraggio clinico. L'uso del farmaco in donne in gravidanza non è stato studiato; pertanto, le pazienti devono informare immediatamente il medico se scoprono di essere incinte.
Dati
Dati dagli studi sugli animali
L'effetto del pegvisomant sullo sviluppo embrionale precoce e sullo sviluppo embrio-fetale è stato valutato in due studi separati su conigli gravidi ai quali è stato somministrato pegvisomant per via sottocutanea alle dosi di 1, 3 e 10 mg/kg/giorno. Non sono state osservate prove di effetti teratogeni associati all'uso di pegvisomant durante l'organogenesi. Alla dose di 10 mg/kg/giorno (6 volte superiore alla dose terapeutica massima nell'uomo, calcolata in base alla superficie corporea) è stato osservato in entrambi gli studi un lieve aumento riproducibile di perdite post-impianto.
Allattamento
Valutazione del rischio
Secondo informazioni limitate da casi clinici riportati in letteratura, il livello di pegvisomant nel latte materno delle donne è risultato inferiore al limite di rilevamento. Non sono disponibili informazioni sull'effetto del farmaco sui neonati allattati al seno né sull'effetto sulle produzione di latte. È necessario considerare i benefici dell'allattamento al seno per lo sviluppo e la salute del neonato, la necessità clinica della madre di ricevere Somavert e qualsiasi effetto avverso potenziale di Somavert sui neonati allattati al seno o sulla malattia di base della madre.
La paziente deve informare il medico se prevede di allattare al seno.
Donne e uomini in età fertile
È opportuno discutere con le pazienti in premenopausa la possibilità di una gravidanza non pianificata, poiché i benefici terapeutici della riduzione dei livelli di ormone della crescita (GH) e della normalizzazione della concentrazione del fattore di crescita simile all'insulina 1 (IGF-1) nelle donne con acromegalia trattate con pegvisomant possono portare a un miglioramento della fertilità.
Capacità di guidare veicoli e di usare macchinari
Non sono stati effettuati studi sull'effetto del farmaco sulla capacità di guidare veicoli o di usare macchinari.
Modalità e dosaggio
Informazioni sul dosaggio
La dose di carico raccomandata del medicinale Somavert è di 80 mg per via sottocutanea sotto supervisione medica. È necessario garantire un'adeguata formazione sulla tecnica di somministrazione delle iniezioni sottocutanee. A partire dal giorno successivo alla somministrazione della dose di carico, il paziente deve ricevere iniezioni sottocutanee di Somavert da 10 mg giornalieri.
Successivamente, la dose deve essere titolata fino al raggiungimento della normalizzazione della concentrazione sierica di IGF-1 (la concentrazione di IGF-1 nel siero deve essere misurata ogni 4-6 settimane). La scelta della dose non deve basarsi sulla concentrazione dell'ormone della crescita né sui segni e sintomi dell'acromegalia. Non è noto se i pazienti che presentano ancora sintomi dopo il raggiungimento di una concentrazione normale di IGF-1 traggano beneficio da un aumento della dose di Somavert.
-
Se la concentrazione di IGF-1 è elevata, la dose del medicinale deve essere aumentata di 5 mg ogni 4-6 settimane.
-
Se la concentrazione di IGF-1 è al di sotto del range normale, la dose del medicinale deve essere ridotta di 5 mg ogni 4-6 settimane.
-
Il livello di IGF-1 deve essere controllato anche quando si passa da uno schema di somministrazione giornaliero della dose richiesta di Somavert in più iniezioni a uno schema giornaliero con somministrazione della dose richiesta in una singola iniezione (vedere la sezione «Proprietà farmacologiche»).
Il range di dosaggio raccomandato è da 10 a 30 mg per via sottocutanea una volta al giorno; la dose massima giornaliera è di 30 mg per via sottocutanea una volta al giorno.
Valutazione dei parametri biochimici di funzionalità epatica prima dell'inizio della terapia con Somavert
Prima di iniziare la terapia con Somavert, è necessario valutare i livelli basali dei parametri di funzionalità epatica del paziente (livelli sierici di alanina aminotransferasi (ALT), aspartato aminotransferasi (AST), bilirubina totale sierica (BTS) e fosfatasi alcalina). Le raccomandazioni per l'inizio della terapia con Somavert in base ai livelli basali di funzionalità epatica e le raccomandazioni per il monitoraggio dei parametri epatici durante la terapia con Somavert sono riportate nella tabella 3 (vedere la sezione «Avvertenze particolari e precauzioni di impiego»).
Procedura per l'iniezione della dose di carico
Le istruzioni riportate di seguito riguardano la ricostituzione della soluzione e la preparazione della dose di carico da 80 mg e sono destinate al personale sanitario. L'operatore sanitario deve ricostituire la polvere liofilizzata in 4 flaconi di Somavert, ciascuno contenente 20 mg di pegvisomant, utilizzando il solvente fornito in dotazione (per preparare la dose di carico da 80 mg sono necessari 4 flaconi di polvere liofilizzata e 4 siringhe da 2,25 ml contenenti il solvente (acqua per preparazioni iniettabili sterile)). L'operatore sanitario deve inoltre eseguire 4 iniezioni della soluzione ricostituita di Somavert, scegliendo il sito di iniezione tra spalla, parte superiore della coscia, addome o natica del paziente (ogni iniezione in un sito diverso).
- Prima della somministrazione della dose di carico, estrarre dal frigorifero la prima confezione (1 flacone di polvere liofilizzata di Somavert contenente 20 mg di pegvisomant e 1 siringa da 2,25 ml contenente il solvente) circa 10 minuti prima dell'orario previsto per l'iniezione.
- Ricostituire il medicinale Somavert, polvere liofilizzata contenente 20 mg di pegvisomant, nel primo flacone utilizzando il solvente. Quando si utilizza il solvente contenuto in una siringa da 2,25 ml, è necessario iniettare lentamente il contenuto della siringa lungo la parete del flacone contenente la polvere liofilizzata di Somavert. Non dirigere il solvente direttamente sulla polvere.
- Non capovolgere né agitare la soluzione, poiché ciò potrebbe causare la denaturazione della proteina pegvisomant. Mescolare lentamente la soluzione con movimenti rotatori per far sciogliere completamente la polvere liofilizzata. Se nella soluzione ricostituita di Somavert si osserva formazione di schiuma, la soluzione è probabilmente deteriorata e quindi non adatta alla somministrazione.
- Prima dell'uso, controllare visivamente la soluzione ricostituita di Somavert per verificare la presenza di particelle estranee o variazioni di colore. La soluzione ricostituita deve essere limpida. Se la soluzione è torbida, non deve essere utilizzata. Dopo la ricostituzione, la soluzione contiene 20 mg di pegvisomant in 1 ml di soluzione.
- Aspirare 1 ml della soluzione ricostituita di Somavert. La soluzione deve essere utilizzata entro 6 ore dalla ricostituzione.
- Eseguire la prima iniezione sottocutanea della soluzione ricostituita di Somavert (20 mg/ml) nella spalla, nella parte superiore della coscia, nell'addome o nella natica del paziente, con un angolo di 90 gradi.
- Ripetere i passaggi da (a) a (d) per ricostituire la soluzione per la seconda e le successive terza e quarta dose di Somavert da 20 mg.
- Successivamente, eseguire la seconda (e poi la terza e la quarta) iniezione sottocutanea della soluzione ricostituita di Somavert (20 mg/ml) nella spalla, nella parte superiore della coscia, nell'addome o nella natica del paziente (scegliendo un sito diverso rispetto alle iniezioni precedenti), con un angolo di 90 gradi.
- Ripetere i passaggi da (a) a (d) per ricostituire la soluzione per la seconda e le successive terza e quarta dose di Somavert da 20 mg.
- Eseguire la prima iniezione sottocutanea della soluzione ricostituita di Somavert (20 mg/ml) nella spalla, nella parte superiore della coscia, nell'addome o nella natica del paziente, con un angolo di 90 gradi.
- Aspirare 1 ml della soluzione ricostituita di Somavert. La soluzione deve essere utilizzata entro 6 ore dalla ricostituzione.
- Prima dell'uso, controllare visivamente la soluzione ricostituita di Somavert per verificare la presenza di particelle estranee o variazioni di colore. La soluzione ricostituita deve essere limpida. Se la soluzione è torbida, non deve essere utilizzata. Dopo la ricostituzione, la soluzione contiene 20 mg di pegvisomant in 1 ml di soluzione.
- Non capovolgere né agitare la soluzione, poiché ciò potrebbe causare la denaturazione della proteina pegvisomant. Mescolare lentamente la soluzione con movimenti rotatori per far sciogliere completamente la polvere liofilizzata. Se nella soluzione ricostituita di Somavert si osserva formazione di schiuma, la soluzione è probabilmente deteriorata e quindi non adatta alla somministrazione.
- Ricostituire il medicinale Somavert, polvere liofilizzata contenente 20 mg di pegvisomant, nel primo flacone utilizzando il solvente. Quando si utilizza il solvente contenuto in una siringa da 2,25 ml, è necessario iniettare lentamente il contenuto della siringa lungo la parete del flacone contenente la polvere liofilizzata di Somavert. Non dirigere il solvente direttamente sulla polvere.
Procedura per l'iniezione della dose di mantenimento
Le raccomandazioni per il paziente o per la persona che se ne prende cura riguardo alla ricostituzione della soluzione e alla somministrazione delle dosi giornaliere (da 10 a 30 mg) sono riportate di seguito nella «Guida passo dopo passo per la somministrazione del medicinale Somavert».
- Prima della somministrazione della dose, estrarre dal frigorifero 1 flacone di polvere liofilizzata di Somavert contenente 10 mg, 15 mg, 20 mg o 30 mg di pegvisomant e una siringa da 2,25 ml contenente il solvente, circa 10 minuti prima dell'orario previsto per l'iniezione.
- Ricostituire la polvere liofilizzata del medicinale Somavert utilizzando il solvente. Quando si utilizza il solvente contenuto in una siringa da 2,25 ml, è necessario iniettare lentamente il contenuto della siringa lungo la parete del flacone contenente la polvere liofilizzata di Somavert. Non dirigere il solvente direttamente sulla polvere.
- Non capovolgere né agitare la soluzione, poiché ciò potrebbe causare la denaturazione della proteina pegvisomant. Mescolare lentamente la soluzione con movimenti rotatori per far sciogliere completamente la polvere liofilizzata. Se nella soluzione ricostituita di Somavert si osserva formazione di schiuma, la soluzione è probabilmente deteriorata e quindi non adatta alla somministrazione.
- Prima dell'uso, controllare visivamente la soluzione ricostituita di Somavert per verificare la presenza di particelle estranee o variazioni di colore. La soluzione ricostituita deve essere limpida. Se la soluzione è torbida, non deve essere utilizzata. Dopo la ricostituzione, la soluzione contiene 10 mg, 15 mg, 20 mg o 30 mg di pegvisomant in 1 ml di soluzione.
- Aspirare 1 ml della soluzione ricostituita di Somavert. La soluzione deve essere utilizzata entro 6 ore dalla ricostituzione.
Eseguire l'iniezione sottocutanea della soluzione ricostituita di Somavert nella spalla, nella parte superiore della coscia, nell'addome o nella natica con un angolo di 90 gradi.
Passo 1. Materiale necessario
Una confezione di Somavert contiene un flacone con la polvere del medicinale Somavert, una siringa pre-riempita e un ago sicuro.
Saranno inoltre necessari: un batuffolo di cotone, una salvietta alcolica, un contenitore per lo smaltimento degli oggetti taglienti.
| fermaglio per le dita sulla siringa |
| tappo del flacone (con il tappo del flacone rimosso) |
| cappuccio dell'ago |
| Agozzo sicuro |
| Siringa |
| cappuccio protettivo dell'ago |
| stelo dello stantuffo della siringa |
| punta della siringa |
| cilindro della siringa |
| cappuccio della siringa |
| data di scadenza |
| tappo del flacone |
| apertura nel tappo |
| Flacone |
Passaggio 2. Preparazione
Prima di iniziare:
- Mescolare Somavert e il liquido solo quando si è pronti ad iniettare la dose.
- Estrarre dal frigorifero una confezione di Somavert e lasciare che il medicinale raggiunga la temperatura ambiente in un luogo sicuro per almeno 10 minuti prima dell’uso.
- Non riscaldare la confezione di Somavert utilizzando fonti di calore come acqua calda o forno a microonde. Il medicinale deve riscaldarsi naturalmente.
- Lavare le mani con acqua e sapone e asciugarle completamente.
- Aprire la confezione della siringa e dell’ago sicuro in modo da poter prendere comodamente ogni componente necessario durante la preparazione dell’iniezione.
- Non utilizzare siringhe o flaconi se:
- sono danneggiati o difettosi;
- la data di scadenza è trascorsa;
- sono stati congelati, anche se ora sono scongelati (vale solo per le siringhe).
Passaggio 3. Scelta del sito di iniezione
| Choisissez le site d'injection |
| Vita Mantenere almeno 5 cm dal centro dell'ombelico |
| Femori |
| Spalle o parte bassa della schiena Faccia posteriore della parte superiore del braccio (solo per il medico o l'assistente) |
- Per ogni iniezione, scegliere un punto diverso all’interno delle aree designate per le iniezioni.
- Evitare le zone vicine alle ossa o aree con ematomi, arrossamenti, dolore o indurimenti, nonché zone con cicatrici o malattie della pelle.
- Pulire il sito di iniezione con una salvietta imbevuta di alcol.
- Lasciare asciugare il sito di iniezione.
Passaggio 4. Rimozione del tappo dal flacone
| Rimozione del tappo dal flacone |
- Togliere il tappo dal flacone.
- Gettare il tappo. Non sarà più necessario.
Attenzione! Non toccare il setto del flacone con alcunché.
Passaggio 5. Rimozione del cappuccio dalla siringa
| Rimozione del cappuccio dalla siringa |
| Cliccare |
- Rimuovere il tappo dalla siringa, lasciando al suo posto l'ago della siringa. Potrebbe essere necessario esercitare una forza maggiore di quanto sembri a prima vista per rimuovere il tappo.
- Gettare via il tappo della siringa. Non sarà più necessario.
- Tenere la siringa in posizione verticale per evitare perdite di liquido.
Attenzione! Non toccare l'estremità dell'ago su alcuna superficie una volta rimosso il tappo.
Passo 6. Collegamento dell'ago sicuro
| Attacco |
- Premere e ruotare l'ago di sicurezza sulla siringa fino all'arresto.
Passaggio 7. Rimozione del cappuccio dell'ago
| Rimozione del cappuccio dell'ago |
- Rimuovere la protezione dell'ago dalla direzione del tappo dell'ago.
- Rimuovere con attenzione il tappo dell'ago.
- Gettare via il tappo dell'ago. Non sarà più necessario.
Attenzione! Non toccare l'ago con nulla.
Passo 8. Inserimento dell'ago
| Inserimento dell'ago |
- Forare il tappo del flacone al centro con l'ago, come mostrato in figura.
- Sostenere la siringa mentre l'ago è nel tappo del flacone per evitare che l'ago si pieghi.
Passo 9. Aggiunta del liquido
| Aggiunta di liquido |
- Inclinare il flacone e la siringa ad un angolo, come mostrato in figura.
- Premere lentamente lo stantuffo della siringa finché tutto il liquido non sarà passato nel flacone.
- Attenzione! Non dirigere il getto di liquido direttamente sulle polvere. Ciò potrebbe causare la formazione di schiuma, rendendo il medicinale inadatto all'uso.
- Non rimuovere ancora l'ago.
Passo 10. Mescolare la soluzione con movimenti circolari
| Mescolare la soluzione con movimenti circolari |
- Tenere la siringa e il flaconcino in una mano, come mostrato in figura.
- Mescolare con attenzione e lentamente la soluzione, muovendo il flaconcino in cerchio su una superficie piana.
- Continuare a mescolare il liquido con movimenti circolari fino a quando tutto il contenuto in polvere non si sarà completamente disciolto.
Nota. Questo processo può richiedere fino a 5 minuti. Non agitare.
Passo 11. Controllo della soluzione
| Controllo della soluzione |
- Tenendo l’ago nella fiala, osservare attentamente la soluzione. Deve essere limpida e non contenere particelle estranee.
- Non utilizzare il medicinale se:
- la soluzione è torbida o non limpida;
- la soluzione è colorata in qualsiasi tonalità;
- sono presenti particelle o schiuma nella fiala.
Passaggio 12. Spostamento dell’ago
| Spostamento dell'ago |
- Capovolgere il flacone in modo da poter vedere l'apertura nel tappo, come mostrato nella figura.
- Posizionare l'ago verso il basso in modo che la punta dell'ago si trovi nel punto più basso del liquido. In questo modo sarà possibile aspirare dal flacone la maggior quantità possibile di liquido.
- Assicurarsi che lo stantuffo della siringa non si sia spostato. Se lo stantuffo della siringa si è spostato, reinserirlo nella siringa. Ciò consente di rimuovere tutta l'aria dalla siringa prima di iniziare a prelevare la dose.
Passo 13. Prelievo della dose
| Titolazione del dosaggio |
- Tirare lentamente il pistone della siringa per aspirare nel cilindro la massima quantità possibile di soluzione dal flacone.
Nota: se si notano bolle d'aria nella siringa, bussare delicatamente sul cilindro della siringa in modo che le bolle risalgano in superficie, quindi espellerle con attenzione nel flacone.
- Rimuovere l'ago dal flacone.
Passaggio 14. Introduzione dell'ago
| Inserimento dell'ago |
- Prendere con attenzione una piega della pelle nel sito di iniezione.
- Inserire l'ago per tutta la sua lunghezza nella piega della pelle.
Passaggio 15. Esecuzione dell'iniezione del medicinale
| Esecuzione dell'iniezione del farmaco |
- Spingere lentamente lo stantuffo della siringa verso il basso fino a quando il cilindro della siringa non sarà vuoto.
Nota. Assicurarsi di aver inserito l'ago per tutta la sua lunghezza.
- Rilasciare la piega della pelle ed estrarre l'ago perpendicolarmente.
Passaggio 16. Sicurezza nell'uso dell'ago
| Clic! |
| Sicurezza dell'ago |
- Abbassi con attenzione il cappuccio protettivo sull'ago.
Con attenzione, prema sul cappuccio utilizzando una superficie rigida per riportarlo nella posizione iniziale.
- Nota. Sentirà un clic quando il cappuccio protettivo andrà a scattare al suo posto.
Passaggio 17. Smaltimento
- Riponga le siringhe utilizzate in un contenitore apposito per lo smaltimento di oggetti taglienti.
- Non getti (non smaltisca) le siringhe con i rifiuti domestici.
Passaggio 18. Dopo l'iniezione
- Se necessario, prenda un batuffolo di cotone pulito e prema delicatamente sulla sede dell'iniezione.
- Non strofinare la sede dell'iniezione.
Bambini.
La sicurezza e l'efficacia dell'uso di Somavert nei bambini non sono state stabilite.
Sovradosaggio.
Durante studi clinici post-marketing è stato riportato un caso di sovradosaggio acuto con Somavert: un paziente ha effettuato autonomamente iniezioni alla dose di 80 mg/giorno (pari a 2,7 volte la dose di mantenimento massima raccomandata) per sette giorni. Nel paziente è stata osservata una lieve aumento della stanchezza, senza altre lamentele né significative alterazioni nei risultati degli esami ematochimici clinici.
In caso di sovradosaggio, l'assunzione di Somavert deve essere interrotta e non deve essere ripresa finché il livello di IGF-1 non sia tornato alla normalità.
Effetti indesiderati.
Gli effetti indesiderati clinicamente rilevanti menzionati nella sezione "Speciali avvertenze e precauzioni per l'uso" sono i seguenti:
- ipoglicemia associata alla riduzione dei livelli dell'ormone della crescita in pazienti con diabete mellito;
- epatotossicità;
- reattività incrociata con i test quantitativi per l'ormone della crescita;
- lipohipertrofia;
- reazioni sistemiche di ipersensibilità.
Durante gli studi clinici pre-approvazione, un aumento della concentrazione sierica di ALT e AST superiore a 10 volte il LSN è stato osservato in due pazienti (0,8%) trattati con Somavert. In un paziente è stata effettuata una nuova prova di provocazione con Somavert, dopo la quale si è verificato un recidiva dell'aumento delle transaminasi, indicando una probabile relazione causale tra la somministrazione del farmaco e l'aumento degli enzimi epatici. I risultati della biopsia epatica eseguita sul secondo paziente erano compatibili con epatite cronica di eziologia sconosciuta. In entrambi i pazienti, i livelli elevati di transaminasi sono tornati alla normalità dopo l'interruzione del trattamento.
L'aumento dei livelli di ALT e AST non è stato accompagnato da un aumento della bilirubina totale e della fosfatasi alcalina, ad eccezione di due pazienti con un lieve aumento associato della fosfatasi alcalina (cioè inferiore a 3 × LSN). L'aumento delle transaminasi è probabilmente indipendente dalla dose di Somavert somministrata e si verifica solitamente tra le 4 e le 12 settimane dall'inizio della terapia. Non sono stati identificati fattori prognostici biochimici, fenotipici o genetici associati a questo fenomeno.
Esperienza dagli studi clinici
Poiché gli studi clinici vengono condotti in condizioni molto diverse, la frequenza degli effetti indesiderati osservati negli studi clinici di un farmaco non può essere direttamente confrontata con quella di un altro farmaco né si può prevedere la stessa frequenza nell'uso clinico reale.
In uno studio randomizzato, in doppio cieco, controllato con placebo, della durata di 12 settimane, con dosi fisse di Somavert in pazienti con acromegalia, 32 pazienti hanno ricevuto placebo e 80 pazienti hanno ricevuto Somavert una volta al giorno (vedi sezione "Proprietà farmacologiche" ("Studi clinici")). In totale, 108 pazienti (30 nel gruppo placebo, 78 nel gruppo Somavert) hanno completato le 12 settimane di terapia.
In totale, otto pazienti con acromegalia (5,3%) sono stati ritirati dagli studi clinici pre-approvazione a causa di effetti indesiderati, inclusi due pazienti con marcato aumento dei livelli di transaminasi, un paziente con lipohipertrofia nei siti di iniezione e un paziente con significativo aumento di peso corporeo. La maggior parte degli effetti indesiderati sembra non essere dipendente dalla dose. Nella Tabella 5 sono riportate le frequenze degli effetti indesiderati segnalati in almeno due pazienti trattati con Somavert e con frequenza superiore rispetto al gruppo placebo nello studio controllato con placebo della durata di 12 settimane.
Tabella 5. Effetti indesiderati osservati nello studio controllato con placebo della durata di 12 settimane in pazienti con acromegalia*
Effetti indesiderati |
Placebo n = 32 |
Somavert |
||
| 10 mg/giorno n = 26 |
15 mg/giorno n = 26 |
20 mg/giorno N = 28 |
||
| Infezione† |
2 (6 %) |
6 (23 %) |
0 |
0 |
| Dolore |
2 (6 %) |
2 (8 %) |
1 (4 %) |
4 (14 %) |
| Nausea |
1 (3 %) |
0 |
2 (8 %) |
4 (14 %) |
| Diarrea |
1 (3 %) |
1 (4 %) |
0 |
4 (14 %) |
| Alterazione dei parametri biochimici della funzionalità epatica |
1 (3 %) |
3 (12 %) |
1 (4 %) |
1 (4 %) |
| Sindrome di tipo influenzale |
0 |
1 (4 %) |
3 (12 %) |
2 (7 %) |
| Reazione nel sito di iniezione |
0 |
2 (8 %) |
1 (4 %) |
3 (11 %) |
| Capogiri |
2 (6 %) |
2 (8 %) |
1 (4 %) |
1 (4 %) |
| Lesione accidentale |
1 (3 %) |
2 (8 %) |
1 (4 %) |
0 |
| Dolore alla schiena |
1 (3 %) |
2 (8 %) |
0 |
1 (4 %) |
| Senite |
1 (3 %) |
2 (8 %) |
0 |
1 (4 %) |
| Dolore al torace |
0 |
1 (4 %) |
2 (8 %) |
0 |
| Edema periferico |
0 |
2 (8 %) |
0 |
1 (4 %) |
| Ipertensione arteriosa |
0 |
0 |
2 (8 %) |
0 |
| Pararestesia |
2 (6 %) |
0 |
0 |
2 (7 %) |
* La tabella include solo le reazioni segnalate in almeno 2 pazienti e osservate più frequentemente nei pazienti trattati con Somavert rispetto ai pazienti del gruppo placebo.
† Nel gruppo trattato con Somavert 10 mg sono state segnalate 6 reazioni codificate con il termine «infezione»: sintomi da raffreddore (3), infezione delle vie respiratorie superiori (1), bolle (1) e infezione dell'orecchio (1). Nel gruppo placebo sono state registrate 2 reazioni di questo tipo: sintomi da raffreddore (1) e infezione del torace (1).
Esperienza post-commercializzazione
Reazioni avverse segnalate spontaneamente dopo l'immissione in commercio
Durante il periodo post-commercializzazione del medicinale Somavert sono state osservate le seguenti reazioni avverse. Poiché queste segnalazioni derivano da fonti spontanee in popolazioni di dimensioni sconosciute, non sempre è possibile valutare in modo affidabile la loro frequenza o stabilire un rapporto causale con l'uso del medicinale.
Durante il periodo post-commercializzazione sono state segnalate reazioni sistemiche di ipersensibilità, comprese reazioni anafilattiche, laringospasmo, angioedema e reazioni cutanee generalizzate (eruzioni, eritema, prurito, orticaria). Alcuni pazienti hanno richiesto il ricovero ospedaliero. Dopo la ripresa del trattamento, i sintomi non si sono ripresentati in nessun paziente (vedere la sezione «Informazioni importanti sull'uso» («Reazioni sistemiche di ipersensibilità»)).
Reazioni avverse segnalate in uno studio osservazionale
ACROSTUDY è un registro osservazionale internazionale che ha raccolto dati sulla sicurezza a lungo termine in 2221 pazienti con acromegalia trattati con Somavert, con una durata media del trattamento di 8,5 anni. Durante il periodo di registrazione, ai pazienti era consentito ricevere anche altre terapie per l'acromegalia. Il dosaggio e lo schema terapeutico erano scelti a discrezione del medico prescrittore. Sebbene il monitoraggio della sicurezza secondo le raccomandazioni fosse obbligatorio, non tutte le valutazioni sono state effettuate in tutti i momenti previsti per ogni paziente. Pertanto, non è possibile confrontare la frequenza delle reazioni avverse con quella osservata negli studi clinici originali.
Tra i 1327 pazienti con livelli normali iniziali di AST e ALT, in 20 (1,5%) si è osservato un aumento dei valori superiore a 3-5 volte il LSN e in 22 (1,7%) un aumento superiore a 5 volte il LSN. La lipohipertrofia è stata segnalata in 35 (1,6%) pazienti.
Tra i 1795 partecipanti allo studio con risultati di risonanza magnetica (RMN) disponibili all'inizio dello studio e almeno un ulteriore esame durante il follow-up, i risultati di RMN hanno mostrato un aumento in 128 (7,1%), una riduzione in 310 (17,3%), sia aumento che riduzione in 81 (4,5%) e nessun cambiamento in 1276 (71,1%).
È necessario informare i pazienti (e/o le persone che li assistono) sulla possibile insorgenza delle seguenti reazioni avverse.
- Le reazioni avverse più frequentemente segnalate sono state reazioni nel sito di iniezione, aumento dei livelli biochimici degli indici di funzionalità epatica, dolore, nausea e diarrea.
- Se nei pazienti si osserva un aumento dei livelli biochimici degli indici di funzionalità epatica, potrebbe essere necessario effettuare prelievi ematici più frequenti per valutare questi parametri e/o interrompere il trattamento con Somavert. Informare i pazienti che devono interrompere immediatamente il trattamento con questo medicinale e contattare il proprio medico in caso di comparsa di ittero.
- Nei pazienti con acromegalia, i tumori secernenti ormone della crescita possono aumentare di dimensioni e devono essere attentamente monitorati mediante risonanza magnetica (RMN).
- Nel sito di iniezione sottocutanea può verificarsi un indurimento che può portare alla formazione di noduli; l'applicazione dell'iniezione in un sito diverso ogni volta può aiutare a prevenire o minimizzare questo fenomeno.
- Nei pazienti con diabete mellito potrebbe essere necessario un attento monitoraggio dello stato clinico e una riduzione della dose di insulina e/o degli agenti ipoglicemizzanti orali durante il trattamento con Somavert.
- Nei pazienti che assumono oppioidi potrebbero essere necessarie dosi più elevate di Somavert per ottenere un adeguato inibizione dell'IGF-1.
Segnalazione delle reazioni avverse sospette
La segnalazione delle reazioni avverse dopo l'autorizzazione all'immissione in commercio è di fondamentale importanza. Permette di continuare a monitorare il rapporto beneficio/rischio del medicinale. Il personale medico e farmaceutico, così come i pazienti o i loro rappresentanti legali, devono segnalare qualsiasi sospetta reazione avversa e la mancata efficacia del medicinale attraverso il sistema informatizzato di farmacovigilanza al seguente indirizzo: https://aisf.dec.gov.ua.
Periodo di validità.
3 anni.
Condizioni di conservazione.
Conservare i flaconcini con il liofilizzato nell'imballaggio originale al riparo dalla luce, a una temperatura compresa tra 2 e 8 °C in frigorifero. Non congelare.
Il medicinale in flaconcini nell'imballaggio originale può essere conservato a una temperatura non superiore a 25 °C per un solo periodo di 30 giorni. Sulla scatola di cartone deve essere indicata la data entro cui il medicinale deve essere utilizzato (entro 30 giorni dalla data di rimozione dal frigorifero). I flaconcini devono essere protetti dalla luce e non devono essere riposti nuovamente in frigorifero. Il medicinale deve essere eliminato se non utilizzato entro 30 giorni di conservazione a temperatura ambiente o alla scadenza indicata sulla scatola, a seconda di quale delle due condizioni si verifichi prima.
Il solvente in siringhe preriempite deve essere conservato a una temperatura non superiore a 30 °C oppure a una temperatura compresa tra 2 e 8 °C in frigorifero. Non congelare.
La soluzione ricostituita deve essere utilizzata immediatamente o entro 6 ore dalla preparazione.
Conservare fuori dalla portata dei bambini.
Confezionamento.
10 flaconcini con liofilizzato in una scatola di cartone intermedia; 3 scatole di cartone intermedie, 30 siringhe preriempite con solvente da 1 ml, 30 aghi sicuri in una scatola di cartone.
Categoria di fornitura.
Sotto prescrizione medica.
Produttore.
Pfizer Manufacturing Belgium NV / Pfizer Manufacturing Belgium NV.
Indirizzo del produttore e sede operativa.
Rijksweg 12, Puurs-Sint-Amands, 2870, Belgio / Rijksweg 12, Puurs-Sint-Amands, 2870, Belgium.