Signifor LAR
UcrainaIndice
ISTRUZIONI PER L'USO MEDICINALE DEL MEDICINALE SIGNIFFOR LAR (SIGNIFOR® LAR®)
Composizione:
Principio attivo: pasireotide;
1 flaconcino con polvere per sospensione iniettabile contiene 20 mg, 40 mg o 60 mg di pasireotide (come pasireotide pamoato).
Eccipienti:
polvere: poli(D,L-lattide-co-glicolide) (50-60:40-50), poli(D,L-lattide-co-glicolide) (50:50);
solvente: 1 siringa preriempita con 2 ml di solvente contiene: carmellosa sodica, mannitolo (E421), polossamero 188, acqua per preparazioni iniettabili.
Forma farmaceutica. Polvere per sospensione iniettabile.
Principali caratteristiche fisico-chimiche:
polvere: polvere da lievemente giallastra a giallastra;
solvente: soluzione trasparente da incolore a giallastra o leggermente brunastra.
Categoria farmacoterapeutica. Ormoni ipofisari, ipotalamici e loro analoghi. Somatostatina e suoi analoghi. Codice ATC H01C B05.
Proprietà farmacodinamiche.
Mecanismo d'azione. Pasireotide è un cipeptides ciclico, un analogo iniettabile della somatostatina. Analogamente agli ormoni peptidici naturali somatostatina-14 e somatostatina-28 (noti anche come fattore inibitorio della secrezione dell'ormone della crescita [SRIF]) e ad altri analoghi della somatostatina, pasireotide esercita il suo effetto farmacologico legandosi ai recettori della somatostatina. Sono noti cinque sottotipi di recettori umani della somatostatina: hsst1, 2, 3, 4 e 5. In condizioni fisiologiche normali, questi sottotipi di recettori sono espressi in diversi tessuti. Gli analoghi della somatostatina si legano ai recettori hsst con diversa potenza (vedi tabella seguente). Pasireotide si lega con elevata affinità a quattro dei cinque recettori hsst.
Affinità di legame della somatostatina (SRIF-14), pasireotide, octreotide e lanreotide ai cinque sottotipi umani di recettori sst (hsst1–5).
| Medicinale |
hsst1 |
hsst2 |
hsst3 |
hsst4 |
hsst5 |
| Somatostatina (SRIF-14) |
0,93 ± 0,12 |
0,15 ± 0,02 |
0,56 ± 0,17 |
1,5 ± 0,4 |
0,29 ± 0,04 |
| Pasireotide |
9,3 ± 0,1 |
1,0 ± 0,1 |
1,5 ± 0,3 |
> 100 |
0,16 ± 0,01 |
| Octreotide |
280 ± 80 |
0,38 ± 0,08 |
7,1 ± 1,4 |
>1000 |
6,3 ± 1,0 |
| Lanreotide |
180 ± 20 |
0,54 ± 0,08 |
14 ± 9 |
230 ± 40 |
17 ± 5 |
I risultati sono espressi come media ± errore standard medio (SEM) del valore IC50, espresso in nmol/l.
Effetti farmacodinamici. I recettori della somatostatina sono espressi in numerosi tessuti, in particolare nei tumori neuroendocrini, nei quali si osserva una secrezione eccessiva di ormoni, tra cui l'ormone della crescita (GH) nell'acromegalia e l'ormone adrenocorticotropo (ACTH) nella malattia di Cushing.
Gli studi in vitro hanno mostrato che le cellule tumorali corticotrope prelevate da pazienti con malattia di Cushing presentano un'espressione marcata di hsst5, mentre i recettari di altri sottotipi sono assenti o espressi in misura trascurabile. Pasireotide si lega e attiva quattro dei cinque hsst, in particolare hsst5, nelle adenomi corticotropi produttori di ACTH, determinando l'inibizione della secrezione di ACTH.
Grazie al suo ampio profilo di legame ai recettori della somatostatina, pasireotide è in grado di stimolare i sottotipi recettoriali hsst2 e hsst5, necessari per inibire la secrezione di GH e del fattore di crescita simile all'insulina (IGF-1), risultando pertanto efficace nel trattamento dell'acromegalia.
Metabolismo del glucosio. In uno studio randomizzato in doppio cieco condotto su volontari sani, l'insorgenza di iperglicemia dopo somministrazione sottocutanea di pasireotide alle dosi di 0,6 mg e 0,9 mg due volte al giorno è stata associata a una riduzione significativa della secrezione di insulina e di incretina (in particolare del peptide glucagone-simile-1 (GLP-1) e del polipeptide insulinotropo dipendente dal glucosio (GIP)). Pasireotide non ha influenzato la sensibilità all'insulina.
Popolazione pediatrica. L'Agenzia europea per i medicinali ha rinunciato all'obbligo di presentare i risultati degli studi sul medicinale Signifor LAR nei bambini di tutte le sottocategorie per acromegalia e gigantismo ipofisario, nonché per malattia di Cushing dipendente dall'ipofisi, ipersecrezione ipofisaria di ACTH e ipercorticosurrenalismo dipendente dall'ipofisi (per informazioni sull'uso nei bambini, vedere la sezione «Modalità di somministrazione e posologia»).
Farmacocinetica.
Pasireotide per somministrazione intramuscolare è disponibile sotto forma di microsfere a rilascio prolungato. Dopo una singola iniezione, la concentrazione plasmatica di pasireotide mostra un iniziale rilascio "a impulso" nel primo giorno dall'iniezione, seguito da una riduzione della concentrazione tra il 2° e il 7° giorno. Successivamente si osserva un graduale raggiungimento della concentrazione massima intorno al 21° giorno e una fase di riduzione lenta nelle settimane successive, insieme alla fase finale di degradazione della matrice polimerica della formulazione.
Assorbimento. La biodisponibilità relativa di pasireotide per via intramuscolare rispetto alla somministrazione sottocutanea è completa. Non sono stati condotti studi volti a valutare la biodisponibilità assoluta di pasireotide nell'uomo.
Distribuzione. In volontari sani, pasireotide somministrato per via intramuscolare si distribuisce ampiamente con un elevato volume di distribuzione (Vz/F > 100 litri). La distribuzione tra cellule del sangue e plasma è indipendente dalla concentrazione e indica che pasireotide si localizza prevalentemente nel plasma (91%). Il legame alle proteine plasmatiche è moderato (88%) e indipendente dalla concentrazione.
Sulla base di dati in vitro, pasireotide è un substrato del trasportatore di efflusso glicoproteina-P. Secondo dati in vitro, pasireotide non è un substrato del trasportatore di efflusso BCRP (proteina di resistenza al cancro della mammella) né dei trasportatori di afflusso OCT1 (trasportatore di cationi organici 1), OATP1B1, 1B3 o 2B1. A dosi terapeutiche, pasireotide non è inoltre un inibitore di UGT1A1, OATP1B1 o 1B3, OAT1 o OAT3, OCT1 o OCT2, P-gp, BCRP, MRP2 o BSEP.
Biotrasformazione. Pasireotide è altamente stabile dal punto di vista metabolico. I dati degli studi in vitro indicano che pasireotide non è un substrato, inibitore o induttore di alcun enzima CYP450 principale. In volontari sani, pasireotide è presente principalmente in forma inalterata nel plasma, nelle urine e nelle feci.
Eliminazione. Pasireotide viene eliminato principalmente attraverso il clearance epatico (escrezione biliare), con un contributo minimo della via renale. In uno studio ADME (assorbimento, distribuzione, metabolismo, eliminazione) nell'uomo, il 55,9 ± 6,63% della dose radioattiva è stato recuperato entro i primi 10 giorni dopo la somministrazione, compreso il 48,3 ± 8,16% di radioattività nelle feci e il 7,63 ± 2,03% nelle urine.
Il clearance (CL/F) di pasireotide intramuscolare in volontari sani è mediamente compreso tra 4,5 e 8,5 l/ora. Sulla base dei risultati dell'analisi farmacocinetica di popolazione, il CL/F misurato è risultato approssimativamente compreso tra 4,8 e 6,5 l/ora nei pazienti con malattia di Cushing tipica e tra 5,6 e 8,2 l/ora nei pazienti con acromegalia tipica.
Linearità e dipendenza dal tempo. Lo stato stazionario farmacocinetico dopo somministrazione intramuscolare di pasireotide viene raggiunto dopo 3 mesi. Dopo diverse somministrazioni mensili nel range da 10 mg a 60 mg ogni 4 settimane, pasireotide per via intramuscolare mostra un effetto farmacocinetico approssimativamente proporzionale alla dose nei pazienti.
Categorie speciali di pazienti.
Popolazione pediatrica. Non sono stati condotti studi su bambini.
Pazienti con compromissione renale. Il clearance renale svolge un ruolo minimo nell'eliminazione di pasireotide nell'uomo. In uno studio clinico, una singola dose sottocutanea di pasireotide 900 mcg in pazienti con disfunzione renale, con compromissione renale lieve, moderata o grave o malattia renale allo stadio terminale (ESRD) non ha avuto un impatto significativo sull'esposizione plasmatica totale a pasireotide. L'esposizione plasmatica a pasireotide non legato (AUCinf,u) è aumentata nei pazienti con compromissione renale (lieve: +33%; moderata: +25%; grave: +99%; ESRD: +143%) rispetto ai pazienti del gruppo di controllo.
Pazienti con compromissione epatica. Non sono stati condotti studi clinici sulla somministrazione intramuscolare di pasireotide in pazienti con compromissione epatica. In uno studio clinico con somministrazione sottocutanea di una singola dose di pasireotide in pazienti con compromissione epatica, sono state osservate differenze statisticamente significative nei pazienti con compromissione epatica moderata e grave (classi B e C secondo Child-Pugh). Nei pazienti con compromissione epatica moderata e grave, l'AUCinf è aumentata rispettivamente del 60% e del 79%, il Cmax del 67% e del 69%, mentre il CL/F è diminuito del 37% e del 44%.
Pazienti anziani (≥ 65 anni). L'età non rappresenta un parametro significativo nell'analisi farmacocinetica di popolazione.
Caratteristiche demografiche. L'analisi farmacocinetica di popolazione della somministrazione intramuscolare di pasireotide indica che la razza e il sesso del paziente non influenzano i parametri farmacocinetici del medicinale. Il peso corporeo ha avuto un lieve impatto sulla farmacocinetica nello studio condotto su pazienti precedentemente non trattati, ma non ha avuto impatto nello studio su pazienti con malattia non adeguatamente controllata. Le donne con acromegalia hanno mostrato un'esposizione maggiore (rispettivamente del 32% e del 51%) rispetto agli uomini negli studi su pazienti non trattati e su pazienti con malattia non adeguatamente controllata; tali differenze non sono risultate clinicamente rilevanti sulla base dei dati di efficacia e sicurezza.
Caratteristiche cliniche.
Indicazioni.
Trattamento di pazienti adulti con acromegaliа per i quali l'intervento chirurgico non è ottimale o è fallito e nei quali non è stato ottenuto un adeguato controllo con un altro analogo della somatostatina.
Trattamento di pazienti adulti con malattia di Cushing per i quali l'intervento chirurgico non è ottimale o è fallito.
La dose di 60 mg è utilizzata soltanto per il trattamento dell'acromegalia.
Controindicazioni.
Ipersensibilità al principio attivo o ad uno qualsiasi degli eccipienti del medicinale. Grave compromissione della funzionalità epatica (classe C secondo Child-Pugh).
Interazioni con altri medicinali e altre forme di interazione.
Interazioni farmacocinetiche previste che determinano un effetto sul pasireotide. L'effetto dell'inibitore della glicoproteina-P verapamil sulla farmacocinetica del pasireotide dopo somministrazione sottocutanea è stato studiato in uno studio sull'interazione tra farmaci con soggetti sani. Non sono state osservate variazioni della farmacocinetica (velocità o entità dell'effetto) del pasireotide.
Interazioni farmacocinetiche previste che determinano un effetto su altri medicinali. Il pasireotide può ridurre la biodisponibilità relativa della ciclosporina. Quando pasireotide e ciclosporina vengono somministrati contemporaneamente, può essere necessario aggiustare la dose di ciclosporina per mantenere una concentrazione terapeutica.
Interazioni farmacodinamiche previste. Medicinali che prolungano l'intervallo QT.
Il pasireotide deve essere somministrato con cautela ai pazienti che assumono contemporaneamente medicinali che prolungano l'intervallo QT, come antiaritmici di classe Ia (ad esempio chinidina, procainamide, disopiramide), antiaritmici di classe III (ad esempio amiodarone, dronedarone, sotalolo, dofetilide, ibutilide), alcuni agenti antibatterici (eritromicina endovenosa, iniezioni di pentamidina, claritromicina, moxifloxacina), alcuni farmaci antipsicotici (ad esempio clorpromazina, tiordazina, flufenazina, pimozide, aloperidolo, tiapride, amisulpride, sertindolo, metadone), alcuni antistaminici (ad esempio terfenadina, astemizolo, mizolastina), farmaci antimalarici (ad esempio clorochina, halofantrina, lumefantrina), alcuni agenti antifungini (ketoconazolo, eccetto lo shampoo).
Medicinali che causano bradicardia. Si raccomanda il monitoraggio clinico del ritmo cardiaco, specialmente all'inizio del trattamento, nei pazienti che ricevono pasireotide contemporaneamente a medicinali che causano bradicardia, come beta-bloccanti (ad esempio metoprololo, carteololo, propranololo, sotalolo), inibitori della colinesterasi (ad esempio rivastigmina, fosfostigmina), alcuni bloccanti dei canali del calcio (ad esempio verapamil, diltiazem, bepridil), alcuni antiaritmici.
Insulina e medicinali antidiabetici. Potrebbe essere necessario aggiustare la dose (riduzione o aumento) di insulina e di medicinali antidiabetici (ad esempio metformina, liraglutide, vildagliptin, nateglinide) quando somministrati contemporaneamente al pasireotide.
Caratteristiche d'uso.
Disturbi del metabolismo del glucosio. Alterazioni dei livelli di glucosio nel sangue sono state spesso osservate in volontari sani e in pazienti dopo il trattamento con pasireotide. Iperglicemia e, più raramente, ipoglicemia sono state osservate in pazienti coinvolti negli studi clinici con pasireotide (vedere la sezione «Effetti indesiderati»).
I pazienti che hanno sviluppato iperglicemia hanno generalmente risposto a un trattamento antidiabetico. Riduzione della dose o interruzione del trattamento con pasireotide a causa di iperglicemia sono stati eventi rari negli studi clinici.
Lo sviluppo di iperglicemia è associato alla riduzione della secrezione di insulina e degli ormoni incretini (in particolare il peptide glucagone-simile 1 (GLP-1) e il polipeptide insulinotropo dipendente dal glucosio (GIP)).
Lo stato glicemico (glicemia a digiuno/plasma/ emoglobina A1c (HbA1c)) deve essere valutato prima dell'inizio del trattamento con pasireotide. Il monitoraggio di glicemia a digiuno/HbA1c durante il trattamento deve essere effettuato secondo le raccomandazioni disponibili. L'autosorveglianza della glicemia nel sangue e/o la valutazione della glicemia a digiuno devono essere effettuate settimanalmente per i primi 3 mesi e successivamente periodicamente, secondo la pratica clinica, nonché durante le prime 4-6 settimane successive a qualsiasi aumento di dose. Inoltre, è necessario effettuare il monitoraggio della glicemia a digiuno a 4 settimane e dell'emoglobina A1c a 3 mesi dopo la fine del trattamento.
Se un paziente sviluppa iperglicemia durante l'assunzione del medicinale Signifor LAR, si raccomanda di iniziare o aggiustare il trattamento antidiabetico secondo le raccomandazioni disponibili per il controllo dell'iperglicemia. Se l'iperglicemia persiste nonostante un trattamento farmacologico adeguato, si deve ridurre la dose di Signifor LAR o interrompere il trattamento (vedere la sezione «Interazioni con altri medicinali ed altre forme di interazione»).
Sono stati riportati casi post-marketing di chetoacidosi con l'uso del medicinale Signifor LAR in pazienti con e senza anamnesi di diabete mellito. I pazienti che sviluppano segni e sintomi compatibili con un grave acidosi metabolica devono essere valutati per la possibile insorgenza di chetoacidosi indipendentemente dalla presenza di diabete mellito in anamnesi.
Nei pazienti con un controllo glicemico inadeguato (definito come valore di HbA1c > 8% nonostante il trattamento antidiabetico) il controllo del diabete e il monitoraggio devono essere intensificati prima e durante la terapia con pasireotide.
Test epatici. Nei pazienti che assumevano pasireotide si è generalmente osservato un lieve aumento transitorio dei livelli delle aminotransferasi. Sono stati inoltre riportati casi rari di aumento concomitante dell'ALP (alanina aminotransferasi) superiore a 3 × LSN (limite superiore della norma) e della bilirubina superiore a 2 × LSN (vedere la sezione «Effetti indesiderati»).
Il monitoraggio della funzionalità epatica successivo deve essere effettuato in base alle indicazioni cliniche. Si raccomanda di controllare la funzionalità epatica prima dell'inizio della somministrazione intramuscolare di pasireotide e dopo 2-3 settimane di terapia, quindi mensilmente per i primi 3 mesi dopo l'inizio del trattamento.
I pazienti con aumento dei livelli di transaminasi richiedono un monitoraggio frequente della funzionalità epatica fino al ritorno dei valori ai livelli precedenti al trattamento. Il trattamento con pasireotide deve essere interrotto se il paziente sviluppa ittero o altri segni di disfunzione epatica clinicamente significativa, in caso di aumento persistente di AST (aspartato aminotransferasi) o ALT di 5 × LSN o superiore, oppure se ALT o AST superano 3 × LSN contemporaneamente a un aumento della bilirubina superiore a 2 × LSN. Dopo l'interruzione del trattamento con pasireotide, i pazienti devono essere monitorati fino alla scomparsa dei sintomi. Il trattamento non deve essere ripreso se si sospetta che i disturbi della funzionalità epatica siano correlati alla pasireotide.
Eventi correlati al sistema cardiovascolare. Sono stati riportati casi di bradicardia durante l'uso di pasireotide. Si raccomanda un attento monitoraggio nei pazienti con malattie cardiache e/o fattori di rischio per bradicardia, come bradicardia clinicamente significativa o infarto miocardico recente in anamnesi, blocchi cardiaci di alto grado, insufficienza cardiaca congestizia (classe III o IV secondo la classificazione NYHA), angina instabile, tachicardia ventricolare persistente, fibrillazione ventricolare. Potrebbe essere necessaria una correzione della dose di farmaci, ad esempio beta-bloccanti, bloccanti dei canali del calcio o farmaci per il controllo dell'equilibrio degli elettroliti.
In due studi condotti su volontari sani è stato dimostrato che la pasireotide prolunga l'intervallo QT nell'ECG. Il significato clinico di questo prolungamento non è noto. Negli studi clinici di fase III in pazienti con acromegalia non sono state osservate differenze clinicamente significative riguardo al prolungamento dell'intervallo QT tra la somministrazione intramuscolare di pasireotide e gli analoghi della somatostatina utilizzati come trattamento attivo di confronto. Tutti gli eventi correlati al prolungamento dell'intervallo QT sono stati temporanei e si sono risolti senza intervento terapeutico.
Nessun episodio di tachicardia ventricolare di tipo torsione di punta è stato osservato in alcuno studio clinico con pasireotide.
La pasireotide deve essere utilizzata con cautela e tenendo conto del rapporto beneficio/rischio nei pazienti con fattori di rischio per il prolungamento dell'intervallo QT, come:
- sindrome congenita da prolungamento dell'intervallo QT;
- malattie cardiache non controllate o significative, inclusi infarto miocardico recente, insufficienza cardiaca congestizia, angina instabile o bradicardia clinicamente significativa;
- assunzione concomitante di farmaci antiaritmici o altri medicinali noti per prolungare l'intervallo QT;
- ipokaliemia e/o ipomagnesiemia.
Prima dell'inizio della terapia con il medicinale Signifor LAR si raccomanda di effettuare un ECG basale. È consigliabile effettuare il monitoraggio dell'effetto sull'intervallo QTc a 21 giorni dall'inizio del trattamento e successivamente in base alle indicazioni cliniche. L'ipokaliemia e/o l'ipomagnesiemia devono essere corrette prima dell'inizio del trattamento con Signifor LAR e deve essere effettuato un monitoraggio periodico adeguato durante la terapia.
Ipercortisolismo. L'inibizione della secrezione di ACTH (ormone adrenocorticotropo) può portare a ipocortisolismo in pazienti trattati con Signifor LAR. È pertanto necessario effettuare un monitoraggio e istruire i pazienti sui segni e sintomi associati all'ipocortisolismo (ad esempio debolezza, affaticamento, anoressia, nausea, vomito, ipotensione arteriosa, iperkaliemia, iponatriemia, ipoglicemia). In caso di ipocortisolismo confermato, potrebbe rendersi necessaria una terapia sostitutiva temporanea con steroidi esogeni (glucocorticoidi) e/o una riduzione della dose o una pausa nel trattamento con Signifor LAR. Una rapida riduzione dei livelli di cortisolo può essere associata a una diminuzione del numero di leucociti.
Colecisti e fenomeni correlati. La litiasi biliare (calcolosi biliare) è una reazione avversa nota associata all'uso di analoghi della somatostatina ed è stata spesso riportata negli studi clinici con pasireotide. Sono stati riportati casi post-marketing di colangite con l'uso del medicinale Signifor LAR, la maggior parte dei quali segnalati come complicanze della calcolosi biliare. Si raccomanda pertanto un'ecografia della colecisti prima e a intervalli di 6 e 12 mesi durante la terapia con Signifor LAR. La presenza di calcoli biliari nei pazienti che assumono Signifor LAR ha generalmente un decorso asintomatico; la presenza di calcoli biliari sintomatici deve essere trattata secondo gli standard della pratica clinica.
Ormoni ipofisari. Poiché l'azione farmacologica della pasireotide imita quella della somatostatina, non si può escludere la possibilità di inibizione degli ormoni ipofisari, oltre all'ormone della crescita (GH) e/o IGF-1, nei pazienti con acromegalia e ACTH/cortisolo nei pazienti con malattia di Cushing. È pertanto necessario effettuare il monitoraggio della funzionalità ipofisaria (ad esempio livelli di TSH/livello libero di T4, ormone della crescita) prima e periodicamente durante la terapia con Signifor LAR, secondo gli standard clinici.
Effetto sulla funzione riproduttiva nelle donne. L'effetto terapeutico della riduzione dei livelli di ormone della crescita e della normalizzazione della concentrazione del fattore di crescita simile all'insulina 1 (IGF-1) nelle donne con acromegalia e la riduzione o normalizzazione dei livelli di cortisolo nel siero sanguigno nelle donne con malattia di Cushing potrebbe potenzialmente ripristinare la funzione riproduttiva. Alle pazienti in età fertile deve essere raccomandato di utilizzare un metodo contraccettivo adeguato, se necessario, durante il trattamento con Signifor LAR.
Alterazioni della coagulazione. Pazienti con valori marcatamente aumentati del tempo di protrombina (PT) e del tempo di tromboplastina parziale (PTT), o pazienti in trattamento con derivati della cumarina o anticoagulanti a base di eparina, sono stati esclusi dagli studi clinici con pasireotide, poiché la sicurezza della combinazione con tali anticoagulanti non è stata stabilita. Se l'uso concomitante di derivati della cumarina o anticoagulanti a base di eparina con la somministrazione intramuscolare di Signifor LAR non può essere evitato, ai pazienti deve essere garantito un controllo regolare dei parametri di coagulazione (PT e PTT) e la dose dell'anticoagulante deve essere aggiustata di conseguenza.
Funzionalità renale. A causa dell'aumento dell'esposizione al farmaco non legato, Signifor LAR deve essere utilizzato con cautela nei pazienti con grave compromissione della funzionalità renale o malattia renale allo stadio terminale.
Contenuto di sodio. Questo medicinale contiene meno di 1 mmol di sodio (23 mg) per dose raccomandata, cioè praticamente è privo di sodio.
Uso durante la gravidanza o l'allattamento.
Gravidanza.
I dati sull'uso di pasireotide in donne in gravidanza sono limitati. Gli studi con somministrazione sottocutanea di pasireotide in animali hanno mostrato tossicità riproduttiva. La pasireotide non è raccomandata per l'uso in donne in gravidanza e in donne in età fertile che non utilizzano metodi contraccettivi.
Allattamento.
Non è noto se la pasireotide sia escreta nel latte materno. I dati disponibili sugli studi con somministrazione sottocutanea di pasireotide in ratti hanno mostrato che la pasireotide penetra nel latte. L'allattamento al seno deve essere interrotto durante il trattamento con il medicinale Signifor LAR.
Fertilità.
Gli studi con somministrazione sottocutanea di pasireotide in ratti hanno mostrato effetti sui parametri riproduttivi nelle femmine. L'importanza clinica di questi effetti nell'uomo non è nota.
Capacità di guidare veicoli o di usare macchinari.
Signifor LAR può avere un lieve effetto sulla capacità di guidare veicoli o di usare macchinari. Ai pazienti deve essere raccomandato di prestare cautela quando guidano veicoli o lavorano con macchinari se durante il trattamento con Signifor LAR avvertono affaticamento, capogiri o cefalea.
Modalità e posologia di somministrazione.
Posologia.
Acromegalia.
La dose raccomandata iniziale per il trattamento dell’acromegalia è di 40 mg di pasireotide ogni 4 settimane.
La dose può essere aumentata fino a un massimo di 60 mg nei pazienti in cui i livelli dell’ormone della crescita (GH) e/o del fattore di crescita simile all’insulina 1 (IGF-1) non sono adeguatamente controllati dopo 3 mesi di trattamento con la dose di 40 mg.
In caso di sospette reazioni avverse o di eccessiva risposta al trattamento (IGF-1 < limite inferiore della norma), potrebbe rendersi necessaria una temporanea riduzione della dose. La dose può essere ridotta temporaneamente o in modo permanente.
Malattia di Cushing.
La dose raccomandata iniziale per il trattamento della malattia di Cushing è di 10 mg di pasireotide, somministrati come iniezione intramuscolare profonda ogni 4 settimane.
Dopo il primo mese di trattamento e periodicamente in seguito, i pazienti devono essere sottoposti a valutazione per verificare il beneficio clinico. La titolazione della dose deve essere effettuata ogni 2-4 mesi in base all’efficacia terapeutica e alla tollerabilità. La dose massima di Signifor LAR nella malattia di Cushing è di 40 mg ogni 4 settimane. Se non si osserva beneficio clinico, si dovrà considerare la possibilità di interrompere il trattamento con Signifor LAR.
Il trattamento di sospette reazioni avverse o di un effetto eccessivo del trattamento (livelli di cortisolo < limite inferiore della norma) potrebbe richiedere una riduzione della dose, un’interruzione temporanea o la sospensione del trattamento con Signifor LAR.
Passaggio da somministrazione sottocutanea a intramuscolare nella malattia di Cushing.
Non sono disponibili dati da studi clinici riguardo al passaggio dalla somministrazione sottocutanea a quella intramuscolare di pasireotide. Se tale passaggio è necessario, la dose iniziale raccomandata per il trattamento della malattia di Cushing è di 10 mg di pasireotide come iniezione intramuscolare profonda ogni 4 settimane. È necessario monitorare il paziente per valutare l’efficacia e la tollerabilità del trattamento; potrebbe rendersi necessaria un’ulteriore correzione della dose.
Salto della dose.
Se una dose di Signifor LAR viene saltata, l’iniezione mancata deve essere somministrata il prima possibile. La dose successiva deve essere somministrata 4 settimane dopo l’iniezione effettuata per ristabilire il programma di somministrazione ogni 4 settimane.
Categorie speciali di pazienti.
Pazienti anziani (≥ 65 anni). I dati sull’uso di Signifor LAR nei pazienti di età pari o superiore a 65 anni sono limitati, ma non vi sono evidenze che indichino la necessità di una correzione della dose in questi pazienti.
Compromissione renale. Non è richiesta alcuna correzione della dose nei pazienti con compromissione renale.
Compromissione epatica. Non è richiesta alcuna correzione della dose nei pazienti con lieve compromissione epatica (classe A secondo Child-Pugh).
La dose iniziale raccomandata nei pazienti con acromegalia e compromissione epatica moderata (classe B secondo Child-Pugh) è di 20 mg ogni 4 settimane. La dose massima raccomandata per questi pazienti è di 40 mg ogni 4 settimane.
Malattia di Cushing: la dose iniziale raccomandata per i pazienti con malattia di Cushing e compromissione epatica moderata (classe B secondo Child-Pugh) è di 10 mg ogni 4 settimane, mentre la dose massima raccomandata per questi pazienti è di 20 mg ogni 4 settimane.
Signifor LAR è controindicato nei pazienti con grave compromissione epatica (classe C secondo Child-Pugh).
Modalità di somministrazione.
Signifor LAR deve essere somministrato mediante iniezione intramuscolare profonda da parte di un operatore sanitario specializzato. La sospensione del medicinale deve essere preparata solo immediatamente prima della somministrazione.
I siti per le iniezioni intramuscolari ripetute devono essere alternati: muscoli glutei destro e sinistro.
Componenti del kit per iniezione:
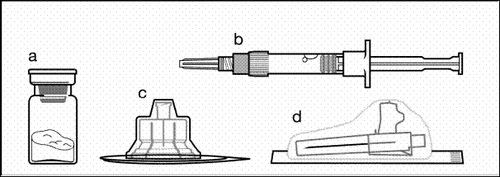
a un flaconcino contenente polvere;
b una siringa preriempita con solvente;
c un adattatore per flaconcino per la preparazione della sospensione;
d un ago sicuro per iniezione (20G × 1,5ʺ).
Per la corretta preparazione della sospensione del medicinale Signifor LAR, è necessario seguire attentamente le istruzioni riportate di seguito.
| Passo 1 Togliere Signifor LAR dal frigorifero. Attenzione Il kit per iniezione deve raggiungere la temperatura ambiente. Lasciare il kit per iniezione a temperatura ambiente per almeno 30 minuti (ma non più di 24 ore). Nota: il kit per iniezione non utilizzato entro 24 ore può essere riposto nuovamente in frigorifero. |
|
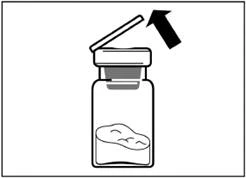
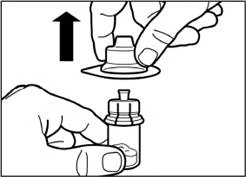
| Passo 2 Rimuovere il coperchio di plastica dal flacone e pulire il tappo di gomma del flacone con un panno alcolico. |
|
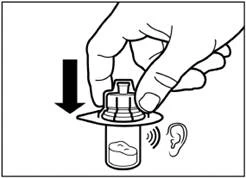
| Rimuovere la pellicola dall'imballo contenente l'applicatore per flacone, ma non estrarre l'applicatore da questo imballo. Tenendo l'applicatore per flacone tramite l'imballo, posizionarlo sulla sommità del flacone e spingere completamente l'applicatore verso il basso fino a quando non scatta in posizione (si sente un caratteristico clic). |
|
| Togliere il contenitore di plastica dall'applicatore per flacone con un movimento verticale verso l'alto. |
|
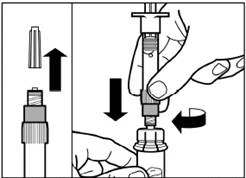
| Passo 3 Rimuovere il tappo dalla siringa precaricata con il solvente e avvitare la siringa sull'adattatore per flaconcino. |
|
| Spingere lentamente lo stantuffo verso il basso fino alla posizione finale per trasferire tutto il solvente nel flaconcino. |
|
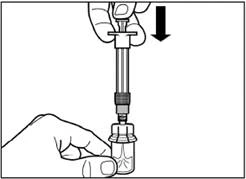
| Passo 4 Attenzione: tenendo premuto lo stantuffo, spostare con cura il flacone in un piano orizzontale per almeno 30 secondi fino a ottenere una sospensione omogenea. Se la polvere non si è completamente sospesa, agitare nuovamente delicatamente il contenuto spostando il flacone in un piano orizzontale per 30 secondi. |
|
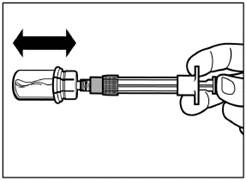
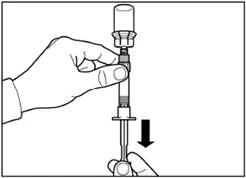
| Passo 5 Rovesciare il flacone con la siringa collegata a testa in giù, lentamente tirare indietro lo stantuffo per trasferire il contenuto dal flacone alla siringa. |
|
| Svitare la siringa dall'adattatore del flacone. |
|
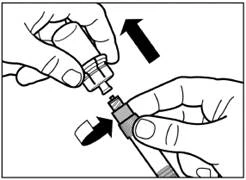
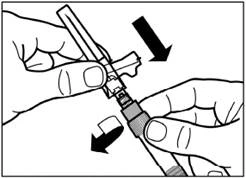
| Passo 6 Avvitare sull'apposita siringa l'ago sicuro per iniezione. |
|
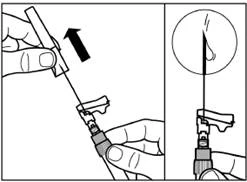
| Rimuovere il tappo protettivo dall'ago tirandolo verso l'alto lungo l'asse dell'ago. Per evitare la sedimentazione, è possibile mantenere l'uniformità della sospensione agitando leggermente la siringa. Battere leggermente sulla siringa in modo che eventuali bolle d'aria visibili risalgano verso l'alto, quindi rimuoverle premendo delicatamente sul pistone. La sospensione è ora pronta per l'uso immediato. |
|
| Passo 7 Signifor LAR può essere somministrato solo per via intramuscolare profonda. Preparare il sito di iniezione disinfettandolo con un batuffolo di alcol. Inserire completamente l'ago nel muscolo gluteo destro o sinistro ad un angolo di 90° rispetto alla superficie della pelle. Tirare leggermente indietro lo stantuffo per accertarsi che l'ago non sia entrato in un vaso sanguigno (se l'ago dovesse entrare in un vaso sanguigno, iniettare in un altro sito). Spingere lentamente lo stantuffo finché la siringa non sarà vuota. Rimuovere l'ago dal sito di iniezione e attivare il meccanismo di protezione (come mostrato nell'immagine qui sotto). |
|
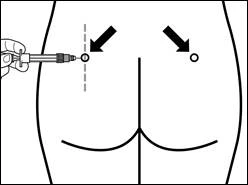
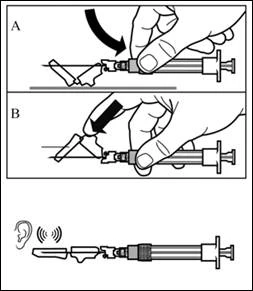
| Passaggio 8 Attivare il meccanismo di protezione dell'ago in uno dei seguenti modi:
Un clic udibile conferma l'attivazione corretta del meccanismo di protezione. Smaltire immediatamente il flacone, la siringa e l'ago nel contenitore per oggetti taglienti. |
|
Popolazione pediatrica.
Non sono stati valutati la sicurezza e l'efficacia del medicinale Signifor LAR nei bambini e negli adolescenti (età da 0 a 18 anni). I dati non sono disponibili.
Sovradosaggio.
In caso di sovradosaggio, si raccomanda di iniziare un trattamento di supporto appropriato, stabilito in base alle condizioni cliniche del paziente e da proseguire fino alla scomparsa dei sintomi.
Effetti indesiderati.
Riassunto del profilo di sicurezza. Il profilo di sicurezza dopo somministrazione intramuscolare di pasireotide è coerente con quello degli analoghi della somatostatina, ad eccezione di una maggiore frequenza e gravità di iperglicemia osservate con la somministrazione intramuscolare di pasireotide. Il profilo di sicurezza dopo somministrazione intramuscolare di pasireotide è in gran parte simile tra i diversi indicazioni terapeutiche – acromegalia e malattia di Cushing.
Acromegalia. La valutazione della sicurezza in acromegalia è stata effettuata su 491 pazienti che hanno ricevuto pasireotide (419 pazienti hanno ricevuto pasireotide per via intramuscolare e 72 per via sottocutanea) durante studi di Fase I, II e III.
Gli effetti indesiderati più comuni (frequenza ≥ 1/10), basati sui dati aggregati di sicurezza degli studi di Fase III C2305 e C2402, sono stati (in ordine decrescente): diarrea (osservata più frequentemente nello studio C2305), calcolosi biliare, iperglicemia (osservata più frequentemente nello studio C2402) e diabete mellito. Gli effetti indesiderati di grado 3 e 4 secondo i criteri comuni di tossicità (CCT) erano principalmente correlati all'iperglicemia.
Malattia di Cushing. Nella malattia di Cushing, la valutazione della sicurezza del farmaco per somministrazione intramuscolare si è basata sui dati raccolti da 150 pazienti trattati con pasireotide nello studio di Fase III G2304 (durata media dell'esposizione 57 settimane). I pazienti sono stati randomizzati in rapporto 1:1 per ricevere dosi iniziali di 10 mg o 30 mg di pasireotide, con possibilità di aumento della dose fino alla dose massima di hidden 40 mg ogni 28 giorni. Gli effetti indesiderati più comuni (frequenza ≥ 1/10) durante lo studio di Fase III G2304 sono stati iperglicemia, diarrea, litiasi biliare e diabete mellito. Frequenza e gravità degli effetti indesiderati sono aumentate con l'aumento della dose iniziale a 30 mg, ma non in modo uniforme per tutti gli effetti indesiderati.
Gli effetti indesiderati includono eventi riportati durante gli studi di base con somministrazione intramuscolare di pasireotide a pazienti con acromegalia e malattia di Cushing. Gli effetti indesiderati sono elencati secondo le classi principali di sistemi e organi del Medical Dictionary for Regulatory Activities (MedDRA). All'interno di ogni classe di sistemi e organi, le reazioni avverse sono ordinate per frequenza. All'interno di ogni gruppo per frequenza, le reazioni avverse sono elencate in ordine decrescente di gravità. La frequenza è definita come segue: molto comune (≥ 1/10); comune (da ≥ 1/100 a < 1/10); non comune (da ≥ 1/1000 a < 1/100); non nota (non può essere stimata a causa della limitatezza dei dati disponibili).
Reazioni avverse (secondo i termini preferenziali) con somministrazione intramuscolare di pasireotide
Patologie del sistema emolinfopoietico: comune – anemia.
Patologie del sistema endocrino: comune – insufficienza surrenalica*.
Disturbi del metabolismo e della nutrizione: molto comune – iperglicemia, diabete mellito; comune – diabete mellito di tipo II, alterata tolleranza al glucosio, diminuzione dell'appetito; non noto – chetoacidosi diabetica.
Patologie del sistema nervoso: comune – cefalea, capogiri.
Patologie cardiache: comune – bradicardia sinusale**, allungamento dell'intervallo QT.
Patologie gastrointestinali: molto comune – diarrea, nausea, dolore addominale***; comune – meteorismo, vomito; non noto – steatorrea, feci acoliche.
Patologie epatobiliari: molto comune – litiasi biliare; comune – colecistite***, colestasi.
Patologie della cute e del tessuto sottocutaneo: comune – alopecia, prurito.
Patologie generali e condizioni inerenti alla sede di somministrazione: molto comune – affaticamento***; comune – reazioni in sede di iniezione***.
Esami diagnostici: comune – aumento dell'emoglobina glicata, aumento dell'alanina aminotransferasi, aumento dell'aspartato aminotransferasi, aumento della gamma-glutammiltransferasi, aumento della glicemia, aumento della glicemia e della creatinfosfocinasi ematica, aumento della lipasi ematica; non comune – aumento dell'amilasi, allungamento del tempo di protrombina.
* L'insufficienza surrenalica comprende insufficienza surrenalica e riduzione dei livelli ematici di cortisolo.
** La bradicardia sinusale comprende bradicardia e bradicardia sinusale.
*** Il dolore addominale comprende dolore addominale e dolore nell'alto dell'addome. Le reazioni in sede di iniezione comprendono dolore in sede di iniezione, formazione di nodulo in sede di iniezione, disagio in sede di iniezione, comparsa di ematoma in sede di iniezione, prurito in sede di iniezione, reazioni in sede di iniezione, ipersensibilità in sede di iniezione e gonfiore in sede di iniezione. La colecistite comprende colecistite acuta e cronica. L'affaticamento comprende affaticamento e astenia.
Descrizione di specifiche reazioni avverse.
Disturbi del metabolismo del glucosio.
Acromegalia. Nei pazienti con acromegalia, l'aumento dei livelli di glucosio a digiuno è stato l'evento di laboratorio di grado 3/4 più comune nei due studi di Fase III. Nello studio C2305, aumenti di glucosio a digiuno di grado 3 sono stati osservati nel 9,7% e 0,6%, e di grado 4 nel 0,6% e 0% dei pazienti con acromegalia trattati rispettivamente con pasireotide intramuscolare e octreotide intramuscolare. Nello studio C2402, aumenti di glucosio a digiuno di grado 3 sono stati riportati nel 14,3% e 17,7% dei pazienti con acromegalia trattati rispettivamente con pasireotide 40 mg e 60 mg intramuscolare, e nello 0% dei pazienti nel gruppo di controllo attivo. Due eventi gravi correlati all'iperglicemia (chetoacidosi diabetica e coma iperglicemico diabetico) sono stati registrati dopo aumento della dose di pasireotide a 60 mg in pazienti precedentemente non trattati: uno in un paziente con iperglicemia non trattata e HbA1c > 8% prima dell'inizio del pasireotide e uno in un paziente con iperglicemia non trattata e glicemia plasmatica a digiuno di 359 mg/dl. In entrambi gli studi, i livelli di FPG e HbA1c sono aumentati nei primi tre mesi di somministrazione intramuscolare di pasireotide. Nei pazienti precedentemente non trattati (studio C2305), l'aumento medio assoluto di FPG e HbA1c è stato generalmente simile in tutti i pazienti trattati con pasireotide intramuscolare, indipendentemente dai valori basali.
Il grado e la frequenza di iperglicemia osservati nei due studi principali con pazienti con acromegalia sono stati più elevati nel gruppo con somministrazione intramuscolare di Signifor LAR rispetto al gruppo di controllo attivo (octreotide per somministrazione intramuscolare o lanreotide come iniezione sottocutanea profonda). Nell'analisi aggregata dei due studi principali, la frequenza totale di eventi avversi correlati all'iperglicemia è stata del 58,6% (tutti i gradi) e del 9,9% (Criteri di Tossicità Comune grado 3 e 4) nel gruppo con somministrazione intramuscolare di Signifor LAR, rispetto al 18,0% (tutti i gradi) e 1,1% (CTC grado 3 e 4) nel gruppo di controllo attivo. In uno studio di riferimento con pazienti non adeguatamente controllati con un altro analogo della somatostatina, la percentuale di pazienti precedentemente non trattati con antidiabetici e che hanno richiesto terapia antidiabetica durante lo studio è stata del 17,5% e 16,1% nei gruppi del farmaco Signifor LAR 40 mg e 60 mg, rispetto all'1,5% nel gruppo di controllo attivo; in uno studio di riferimento con pazienti non precedentemente trattati, la percentuale di pazienti che hanno richiesto terapia antidiabetica durante lo studio è stata del 36% nel gruppo del farmaco Signifor LAR rispetto al 4,4% nel gruppo di controllo attivo.
Malattia di Cushing. Nei pazienti con malattia di Cushing, l'aumento dei livelli di GPN è stato l'evento di laboratorio più comune di grado 3 secondo i CTC (14,7% dei pazienti) durante lo studio di Fase III G2304; non sono stati osservati eventi di grado 4. L'aumento medio di HbA1c è stato meno pronunciato nei pazienti con glicemia normale all'inclusione rispetto ai pazienti con prediabete o diabete. I livelli medi di GPN sono spesso aumentati nel primo mese di trattamento, per poi ridursi e stabilizzarsi nei mesi successivi. Gli aumenti di GPN e HbA1c erano dose-dipendenti e i valori si riducevano generalmente dopo l'interruzione della somministrazione intramuscolare di pasireotide, ma rimanevano superiori ai valori basali. La frequenza totale di reazioni avverse correlate all'iperglicemia è stata del 75,3% (tutti i gradi) e del 22,7% (grado 3 secondo CTC). Reazioni avverse come iperglicemia e diabete mellito hanno portato all'interruzione dello studio in 3 (2,0%) e 4 pazienti (2,7%).
Gli aumenti dei livelli di glucosio e HbA1c nel plasma a digiuno osservati dopo somministrazione intramuscolare di pasireotide sono stati reversibili dopo l'interruzione del farmaco.
Si raccomanda il monitoraggio della glicemia nei pazienti trattati con Signifor LAR.
Disturbi gastrointestinali. Durante il trattamento con il farmaco Signifor LAR, sono stati spesso riportati disturbi gastrointestinali. Questi eventi erano generalmente di lieve entità, non richiedevano intervento e si risolvevano con la prosecuzione del trattamento. Nei pazienti con acromegalia, i disturbi gastrointestinali sono stati meno frequenti nei pazienti non adeguatamente controllati rispetto ai pazienti precedentemente non trattati.
Reazioni in sede di iniezione. Negli studi di Fase III, le reazioni in sede di iniezione (ad esempio dolore in sede di iniezione, disagio in sede di iniezione) erano prevalentemente di grado 1 o 2. La frequenza di questi eventi è stata più alta nei primi 3 mesi di trattamento. Negli studi con pazienti con acromegalia, le reazioni in sede di iniezione sono state simili con somministrazione intramuscolare di pasireotide e somministrazione intramuscolare di octreotide, e sono state meno frequenti nei pazienti non adeguatamente controllati rispetto ai pazienti precedentemente non trattati.
Prolungamento dell'intervallo QT. Nello studio C2305 con pazienti con acromegalia, la percentuale di pazienti con nuovi allungamenti significativi dell'intervallo QT/QTc è stata simile nei gruppi trattati con pasireotide intramuscolare e octreotide intramuscolare prima della fase incrociata, con alcuni valori anomali significativi. Valori di QTcF > 480 ms sono stati osservati in 3 pazienti rispetto a 2 nel gruppo di pasireotide intramuscolare e octreotide intramuscolare rispettivamente, e prolungamenti di QTcF > 60 ms rispetto al valore basale sono stati riportati in 2 pazienti rispetto a 1 nei rispettivi gruppi. Nello studio C2402, l'unico valore anomalo significativo è stato un QTcF > 480 ms in 1 paziente nel gruppo di pasireotide intramuscolare 40 mg. Durante lo studio G2304 con pazienti con malattia di Cushing, un QTcF > 480 ms è stato osservato in 2 pazienti. Valori di QTcF > 500 ms non sono stati osservati in nessuno degli studi principali.
Enzimi epatici. È stato riportato un aumento transitorio degli enzimi epatici con l'uso di analoghi della somatostatina, osservato anche nei pazienti trattati con pasireotide negli studi clinici. L'aumento è stato principalmente asintomatico, di grado lieve e reversibile con la prosecuzione del trattamento. Alcuni casi di aumento concomitante di ALT > 3 × LSN e bilirubina > 2 × LSN sono stati osservati con somministrazione sottocutanea, ma non nei pazienti trattati con pasireotide intramuscolare. Tutti i casi di aumento concomitante si sono verificati entro 10 giorni dall'inizio del trattamento. I pazienti si sono ripresi senza conseguenze cliniche e i test di funzionalità epatica sono tornati ai valori basali dopo l'interruzione del trattamento.
Si raccomanda il monitoraggio degli enzimi epatici prima e durante il trattamento con il farmaco Signifor LAR secondo le norme cliniche.
Enzimi pancreatici. Nei pazienti trattati con pasireotide negli studi clinici, è stato osservato un aumento asintomatico della lipasi e dell'amilasi. L'aumento è stato prevalentemente di grado lieve e reversibile con la prosecuzione del trattamento. La pancreatite è una reazione avversa potenzialmente correlata all'uso di analoghi della somatostatina, a causa del legame tra calcolosi biliare e pancreatite acuta.
Incompatibilità.
A causa della mancanza di studi di compatibilità, questo medicinale non deve essere miscelato con altri medicinali.
Periodo di validità. 3 anni.
Condizioni di conservazione. Conservare nell'imballaggio originale a una temperatura compresa tra 2-8 ºC. Non congelare. Conservare fuori dalla portata dei bambini.
Confezionamento. Polvere in flacone di vetro marrone da 6 ml, chiuso con tappo in gomma grigio e sigillato con capsula in alluminio del sistema flip-off di colore grigio (per dosaggio da 20 mg), rosso (per dosaggio da 40 mg) o arancione (per dosaggio da 60 mg), in dotazione con: solvente in siringa pre-riempita da 3 ml in vetro incolore con due tappi in gomma grigi, fermo per dita, stantuffo e capsula; un ago e un adattatore, il tutto in una scatola di cartone.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore. Recordati Rare Diseases.
Sede del produttore e indirizzo del luogo di esercizio dell'attività.
Eco River Park, 30 Rue des Peupliers, Nanterre, 92000, Francia;
Immeuble Le Wilson, 70 Avenue du Général de Gaulle, Puteaux, 92800, Francia.