Hemax
Ucraina
Indice
ISTRUZIONI PER L'USO MEDICO DEL MEDICINALE Hemax (Hemax)
Composizione:
Principio attivo: eritropoietina (epoetina alfa);
1 flaconcino contiene eritropoietina (epoetina alfa) 1 000 UI, 2 000 UI, 3 000 UI, 4 000 UI, 10 000 UI, 20 000 UI oppure 40 000 UI;
Eccipienti: mannite (E 421), cloruro di sodio, diidrogenofosfato di sodio diidrato, fosfato di sodio dodecaidrato, albumina umana.
Forma farmaceutica. Liofilizzato per soluzione iniettabile.
Principali caratteristiche fisico-chimiche: polvere bianca, omogenea, compatta.
Gruppo farmacoterapeutico. Agenti che agiscono sul sistema emopoietico. Farmaci antianemici. Altri farmaci antianemici. Eritropoietina.
Codice ATC B03X A01.
Proprietà farmacologiche
Farmacodinamica
Hemax è un medicinale il cui principio attivo è l'epoetina alfa (eritropoietina umana ricombinante, r-HuEPO). L'epoetina è una glicoproteina ottenuta mediante tecnologia del DNA ricombinante contenente 165 aminoacidi. Viene prodotta da cellule di mammifero geneticamente modificate che contengono il gene dell'eritropoietina. Il prodotto è altamente purificato e presenta una sequenza di aminoacidi identica a quella dell'eritropoietina umana naturale.
L'eritropoietina induce l'eritropoiesi stimolando la divisione e la differenziazione delle cellule progenitrici eritroidi, portando così ad un aumento del numero di eritrociti e dell'emocrito. L'eritropoietina stimola inoltre il rilascio dei reticolociti dal midollo osseo nel circolo ematico, dove maturano fino a diventare eritrociti. Normalmente, la concentrazione di eritropoietina nel siero è compresa tra 10 e 30 UI/ml ed è regolata dal livello di ossigenazione dei tessuti. Quando la disponibilità di ossigeno nei tessuti diminuisce, la concentrazione di eritropoietina aumenta da 100 a 1000 volte. Questo fenomeno si osserva anche nelle anemie.
Farmacocinetica
Hemax (principio attivo – epoetina alfa) viene somministrato per via parenterale (sottocutanea o endovenosa). Il livello di reticolociti aumenta entro 7-10 giorni dall'amministrazione del farmaco. Il numero di eritrociti e i valori di ematocrito ed emoglobina aumentano generalmente entro 2-6 settimane dall'amministrazione dell'epoetina alfa. L'entità e la velocità della risposta dipendono dalla dose del farmaco e dalla disponibilità di ferro nel siero. La concentrazione massima nel plasma viene raggiunta entro 15 minuti dopo una singola somministrazione endovenosa e tra 5 e 24 ore dopo una singola somministrazione sottocutanea. Il livello massimo di eritropoietina nel siero può mantenersi da 12 a 16 ore dopo la somministrazione sottocutanea, e l'eritropoietina rimane ancora rilevabile nel siero dopo 24 ore.
Il tempo di dimezzamento dell'epoetina alfa varia da 4 a 13 ore dopo somministrazione endovenosa o sottocutanea. Il tempo di dimezzamento dopo la prima dose è più lungo rispetto a quello osservato dopo due o più settimane di trattamento. Generalmente, entro 24 ore il livello di eritropoietina nel plasma ritorna ai valori basali. Dopo somministrazione sottocutanea, la concentrazione massima nel plasma si osserva tra 5 e 24 ore, mentre la successiva riduzione della concentrazione avviene più lentamente.
Negli studi condotti su volontari sani adulti si è osservato che il tempo di dimezzamento dopo somministrazione endovenosa è del 20% inferiore rispetto a quello osservato nei pazienti con insufficienza renale. Il tempo di dimezzamento dopo somministrazione sottocutanea di Hemax in volontari sani adulti è di 20,8 ± 6,3 ore. Dopo l'interruzione del trattamento con Hemax, l'ematocrito inizia a diminuire entro 2 settimane.
Caratteristiche cliniche.
Indicazioni.
- Trattamento dell'anemia sintomatica associata all'insufficienza renale cronica:
- negli adulti e nei bambini in emodialisi e negli adulti in dialisi peritoneale;
- negli adulti con insufficienza renale non ancora sottoposti a emodialisi, per il trattamento dell'anemia grave di origine renale accompagnata da sintomi clinici.
- Trattamento dell'anemia e riduzione del volume delle emazie necessarie nelle trasfusioni ematiche negli adulti sottoposti a chemioterapia per tumori solidi, linfoma maligno o mieloma multiplo e con alto rischio di trasfusione, valutato in base allo stato generale del paziente (compreso lo stato cardiovascolare, anemia preesistente prima dell'inizio della chemioterapia).
- Hemax è indicato nell'ambito di un programma di predonazione per facilitare la raccolta di sangue autologo in pazienti con anemia moderata (livello di emoglobina 10–13 g/dl (6,2–8,1 mmol/l), assenza di carenza di ferro). Il trattamento deve essere prescritto solo in caso di impossibilità o insufficiente efficacia delle procedure di risparmio ematico (quando un intervento chirurgico maggiore programmato prevede una perdita ematica significativa – 4 o più unità di sangue per le donne e 5 o più unità per gli uomini).
- Hemax è indicato negli adulti senza carenza di ferro prima di un intervento chirurgico ortopedico maggiore programmato ad alto rischio di complicanze trasfusionali, al fine di ridurre l'uso di trasfusioni ematiche allogeniche. L'uso del medicinale deve essere limitato agli adulti con anemia moderata (livello di emoglobina compreso tra 10–13 g/dl) che non partecipano a un programma di raccolta di sangue autologo e con una perdita ematica prevista di entità moderata (900–1800 ml).
- Hemax è indicato per il trattamento dell'anemia sintomatica (livello di emoglobina ≤ 10 g/dl) negli adulti con sindrome mielodisplastica (MDS) primitiva a basso rischio o rischio intermedio-1 e con bassi livelli sierici di eritropoietina (<200 mU/ml).
Controindicazioni.
- Ipertensione arteriosa non controllata;
- sviluppo di aplasia eritroide vera (PRCA) in seguito a trattamento con qualsiasi eritropoietina (vedere sezione «Avvertenze speciali»);
- ipersensibilità all'albumina umana;
- ipersensibilità ai farmaci ottenuti da cellule di mammiferi;
- ipersensibilità al principio attivo o a uno qualsiasi degli eccipienti del medicinale;
- gravi malattie coronariche, periferiche arteriose, carotidee o cerebrovascolari, nonché recente infarto del miocardio o ictus in pazienti sottoposti a chirurgia ortopedica maggiore programmata che non abbiano partecipato a un programma di raccolta di sangue autologo;
- impossibilità di applicare una profilassi antitrombotica adeguata nei pazienti chirurgici;
- controindicazioni legate al programma di raccolta di sangue autologo in pazienti che ricevono epoetina alfa.
Interazioni con altri medicinali e altre forme di interazione.
Non sono state osservate interazioni tra il medicinale Hemax e altri farmaci.
Non esistono dati che indichino che il trattamento con epoetina alfa influisca sul metabolismo di altri medicinali.
Farmaci che inibiscono l'eritropoiesi possono ridurre la risposta al trattamento con epoetina alfa.
Poiché il ciclosporina si lega agli eritrociti, esiste la possibilità di un'interazione farmacologica. Quando Hemax e ciclosporina vengono somministrati contemporaneamente, si raccomanda di monitorare i livelli ematici di ciclosporina e di aggiustare la dose in caso di aumento dell'emocrito.
Non esistono evidenze confermate di interazioni tra epoetina alfa e G-CSF (fattore stimolante le colonie di granulociti) o GM-CSF (fattore stimolante le colonie di granulociti e macrofagi) riguardo alla differenziazione ematologica o alla proliferazione delle cellule tumorali in campioni di biopsia in vitro.
In donne adulte con carcinoma mammario metastatico, la somministrazione sottocutanea di epoetina alfa alla dose di 40000 UI/ml contemporaneamente a trastuzumab alla dose di 6 mg/kg non ha influenzato la farmacocinetica di trastuzumab.
Caratteristiche particolari di utilizzo.
La pressione arteriosa deve essere costantemente monitorata in tutti i pazienti durante il trattamento con Hemax. Il medicinale deve essere usato con cautela nei pazienti con ipertensione non trattata, trattamento inadeguato dell'ipertensione o controllo insufficiente dell'ipertensione. Durante il trattamento con Hemax può rendersi necessario iniziare o intensificare una terapia antipertensiva. Se la pressione non può essere controllata, l'uso dell'epoetina alfa deve essere interrotto.
Sono stati osservati casi di crisi ipertensiva con encefalopatia e convulsioni, che richiedevano immediata valutazione medica e terapia intensiva, anche in pazienti con pressione arteriosa normale o bassa all'inizio del trattamento. Si deve prestare particolare attenzione all'insorgenza di cefalea improvvisa, di tipo emicranico, pulsante.
L'epoetina alfa deve essere usata con cautela nei pazienti con epilessia, storia di convulsioni o condizioni mediche che rappresentano fattori di rischio per lo sviluppo di convulsioni, come infezioni del sistema nervoso centrale o metastasi cerebrali.
L'epoetina alfa deve essere usata con cautela nei pazienti con insufficienza epatica cronica. La sicurezza dell'uso dell'epoetina alfa in questa categoria di pazienti non è stata stabilita.
Nei pazienti che ricevono farmaci stimolanti l'eritropoiesi, si osserva un aumento del rischio di malattie vascolari con complicanze trombotiche, in particolare trombosi venosa e arteriosa ed embolia (inclusi esiti letali), come trombosi venosa profonda, embolia polmonare, trombosi della vena retinica e infarto del miocardio. Sono stati riportati anche casi di ictus (inclusi ictus ischemico, emorragico e attacchi ischemici transitori).
Prima di iniziare il trattamento con epoetina alfa, si devono attentamente valutare i rischi di eventi trombovascolari con complicanze trombotiche e il beneficio atteso dall'uso del medicinale, specialmente nei pazienti con fattori di rischio concomitanti, inclusi obesità e storia di eventi trombovascolari (ad esempio trombosi venosa profonda, embolia polmonare e ictus).
Il livello di emoglobina deve essere attentamente monitorato in tutti i pazienti, a causa del potenziale rischio aumentato di complicanze tromboemboliche e di esito letale nel caso di uso del medicinale a livelli di emoglobina superiori al valore target indicato nelle indicazioni.
Durante il trattamento può verificarsi un lieve aumento dose-dipendente del numero di piastrine entro i limiti normali. Questo parametro tende a diminuire nel corso del trattamento successivo. Sono stati riportati anche casi di trombocitosi. Si raccomanda il monitoraggio regolare del numero di piastrine durante le prime 8 settimane di trattamento.
Il livello di ferritina (o la concentrazione di ferro nel siero) deve essere determinato in tutti i pazienti prima dell'inizio e durante il trattamento con Hemax. Se necessario, devono essere somministrati supplementi di ferro. L'assenza di risposta clinica al trattamento con Hemax richiede l'individuazione di fattori concomitanti, come carenza di ferro, acido folico o vitamina B12, intossicazione da alluminio, infezioni intercorrenti, processi infiammatori o traumi, emolisi, fibrosi del midollo osseo di qualsiasi eziologia.
Tutte le altre cause di anemia (carenza di ferro, acido folico, vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi o fibrosi del midollo osseo di qualsiasi origine) devono essere identificate e trattate prima dell'inizio della terapia con epoetina alfa e prima di decidere di aumentare il dosaggio. Nella maggior parte dei casi, i valori di ferritina nel siero tendevano a diminuire contemporaneamente all'aumento dell'ematocrito.
Tutti i pazienti in trattamento con epoetina alfa devono ricevere un adeguato apporto di ferro (200 mg al giorno per via orale) per tutta la durata della terapia. Per garantire un livello sufficiente di ferro nell'organismo, la somministrazione di preparati di ferro deve iniziare il prima possibile, anche diverse settimane prima dell'inizio del programma di raccolta del sangue autologo.
Per garantire una risposta ottimale al trattamento con epoetina alfa, è necessario assicurare un adeguato apporto di ferro:
- ai pazienti con insufficienza renale cronica si raccomanda l'assunzione di ferro (200–300 mg/giorno per gli adulti e 100–200 mg/giorno per i bambini per via orale, calcolati come ferro elementare) se il livello di ferritina nel siero è inferiore a 100 ng/ml;
- ai pazienti con neoplasie si raccomanda l'assunzione di ferro (200–300 mg/giorno per via orale, calcolati come ferro elementare) se la saturazione della transferrina è inferiore al 20%;
- ai pazienti che partecipano a un programma di raccolta del sangue autologo si raccomanda l'assunzione di ferro (200 mg/giorno per via orale, calcolati come ferro elementare) diverse settimane prima dell'inizio della raccolta del sangue autologo, al fine di raggiungere depositi significativi di ferro prima dell'inizio della terapia e durante il trattamento con epoetina alfa;
- ai pazienti in previsione di interventi ortopedici elettivi estesi si raccomanda l'assunzione di ferro (200 mg/giorno per via orale, calcolati come ferro elementare) durante il trattamento con epoetina alfa. Se possibile, l'assunzione di ferro dovrebbe iniziare prima dell'inizio della terapia con epoetina alfa per raggiungere depositi significativi di ferro nell'organismo.
Molto raramente sono stati riportati lo sviluppo o il peggioramento di una porfiria preesistente in pazienti in trattamento con epoetina alfa. L'epoetina alfa deve essere usata con cautela nei pazienti con porfiria.
Sono stati riportati casi di gravi reazioni avverse cutanee correlate al trattamento con epoetina, inclusi il sindrome di Stevens-Johnson e la necrolisi epidermica tossica, che possono essere potenzialmente letali. I casi più gravi si sono verificati soprattutto durante l'uso di epoetine a rilascio prolungato.
I pazienti devono essere informati sulla possibilità di reazioni avverse cutanee. In caso di comparsa di sintomi di reazioni cutanee avverse, il trattamento con epoetina alfa deve essere immediatamente interrotto e si devono considerare metodi alternativi di trattamento.
Se un paziente sviluppa una grave reazione cutanea durante il trattamento con epoetina, come il sindrome di Stevens-Johnson o la necrolisi epidermica tossica, il trattamento con questo medicinale non deve mai essere ripreso.
Il nome commerciale dei farmaci stimolanti l'eritropoiesi utilizzati nel trattamento deve essere chiaramente indicato nella cartella clinica del paziente. La conversione da un agente stimolante l'eritropoiesi a un altro è possibile solo sotto supervisione medica.
Eritroplasia vera (PRCA).
Sono stati riportati casi di eritroplasia vera mediata da anticorpi (PRCA) dopo mesi o anni di somministrazione sottocutanea di epoetina, prevalentemente in pazienti con insufficienza renale cronica. Sono stati riportati anche casi di eritroplasia vera in pazienti con epatite C in trattamento con interferone e ribavirina contemporaneamente a agenti stimolanti l'eritropoiesi. L'epoetina alfa non è indicata per il trattamento dell'anemia associata all'epatite C.
I pazienti in cui si osserva una perdita improvvisa di efficacia terapeutica (manifestata da una riduzione dell'emoglobina di 1–2 g/dl al mese) con aumento della necessità di trasfusioni devono essere indirizzati per valutare il numero di reticolociti e identificare le cause tipiche di ridotta risposta clinica (carenza di ferro, acido folico, vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi o fibrosi del midollo osseo di qualsiasi origine).
In caso di riduzione paradossale dell'emoglobina e sviluppo di anemia grave associata a basso numero di reticolociti, il trattamento con Hemax deve essere immediatamente interrotto, si deve verificare la presenza di anticorpi contro l'eritropoietina e deve essere effettuato uno studio del midollo osseo per confermare la diagnosi di eritroplasia vera (PRCA).
Non si deve somministrare il trattamento con altri agenti stimolanti l'eritropoiesi, poiché esiste la possibilità di reazione crociata.
Trattamento dell'anemia sintomatica in adulti e bambini con insufficienza renale cronica.
Nei pazienti con insufficienza renale cronica in trattamento con epoetina alfa, il livello di emoglobina deve essere monitorato regolarmente fino al raggiungimento di un livello stabile, quindi periodicamente. L'aumento dell'emoglobina dovrebbe essere di circa 1 g/dl (0,62 mmol/l) al mese e non dovrebbe superare i 2 g/dl (1,25 mmol/l) al mese, al fine di minimizzare il rischio di ipertensione arteriosa.
Nei pazienti con insufficienza renale cronica, il livello raggiunto di emoglobina non deve superare il limite superiore della concentrazione emoglobinica desiderata. Negli studi clinici è stato osservato un aumento del rischio di esiti letali e di gravi reazioni avverse cardiovascolari con l'uso di agenti stimolanti l'eritropoiesi per raggiungere una concentrazione di emoglobina superiore a 12 g/dl (7,5 mmol/l).
Studi clinici controllati non hanno dimostrato vantaggi significativi nell'uso di epoetine a concentrazioni di emoglobina superiori a quelle necessarie per controllare i sintomi dell'anemia e prevenire le trasfusioni.
La dose di Hemax deve essere aumentata con cautela nei pazienti con insufficienza renale cronica, poiché alte dosi cumulative di eritropoietina possono essere associate a un aumento del rischio di mortalità e di gravi eventi cardiovascolari e cerebrovascolari. Nei pazienti con risposta inadeguata al trattamento con epoetine, si dovrebbero considerare altre opzioni per affrontare la scarsa risposta.
Lo stato dei pazienti con insufficienza renale cronica in trattamento con Hemax per via sottocutanea deve essere monitorato regolarmente per la perdita di efficacia del trattamento, definita come riduzione o perdita di risposta al trattamento con epoetina alfa in pazienti che in precedenza avevano risposto alla terapia. La perdita di efficacia si caratterizza per una riduzione persistente del livello di emoglobina indipendentemente dall'aumento della dose di epoetina alfa.
Con regimi di trattamento a intervalli di somministrazione prolungati (somministrazione di epoetina alfa meno di una volta alla settimana), in alcuni pazienti il livello di emoglobina può diminuire; tali pazienti potrebbero necessitare di un aumento della dose. Il livello di emoglobina deve essere monitorato regolarmente.
Nei pazienti in emodialisi sono stati osservati trombosi dello shunt, specialmente in quelli con tendenza all'ipotensione o complicanze delle fistole artero-venose (ad esempio stenosi, aneurismi, ecc.). A tali pazienti si raccomanda la verifica dello shunt e la prevenzione della trombosi, ad esempio con acido acetilsalicilico.
In singoli casi è stata osservata iperkaliemia, sebbene non sia stato stabilito un nesso causale. Nei pazienti con insufficienza renale cronica è necessario monitorare i livelli di elettroliti nel siero. In caso di aumento del potassio ematico, oltre al trattamento appropriato per l'iperkaliemia, si deve considerare la possibilità di sospensione temporanea di Hemax fino al ripristino dei livelli normali di potassio.
A causa dell'aumento dell'ematocrito, i pazienti in emodialisi che ricevono Hemax spesso necessitano di un aumento della dose di eparina durante la dialisi. In caso di eparinizzazione inadeguata, può svilupparsi un'occlusione del sistema di dialisi.
Secondo le informazioni attualmente disponibili, l'uso di Hemax per il trattamento dell'anemia nei pazienti adulti pre-dialisi con insufficienza renale non accelera il progresso dell'insufficienza renale.
Trattamento di pazienti con anemia indotta da chemioterapia.
Nei pazienti con neoplasie in trattamento con epoetina alfa, il livello di emoglobina deve essere monitorato regolarmente fino al raggiungimento di un livello stabile, quindi periodicamente.
Le epoetine sono fattori di crescita che stimolano principalmente la produzione di eritrociti. I recettori dell'eritropoietina sono stati identificati anche sulla superficie di diverse cellule tumorali. Come per altri fattori di crescita, non si può escludere la possibilità che le epoetine stimolino la crescita di alcuni tipi di tumore.
Non si può escludere l'effetto degli agenti stimolanti l'eritropoiesi sulla progressione del tumore o sulla riduzione della sopravvivenza libera da progressione della malattia. Negli studi sull'uso di epoetina alfa e di altri agenti stimolanti l'eritropoiesi sono stati osservati:
- riduzione del controllo loco-regionale nei pazienti con cancro della testa e del collo in evoluzione, in trattamento con radioterapia, quando si mirava a un livello di emoglobina superiore a 14 g/dl (8,7 mmol/l);
- riduzione della sopravvivenza globale e aumento del numero di decessi per progressione della malattia entro 4 mesi nei pazienti con cancro al seno metastatico in chemioterapia, quando si mirava a un livello di emoglobina di 12–14 g/dl (7,5–8,7 mmol/l);
- aumento del rischio di esito letale quando si mirava a un livello di emoglobina di 12 g/dl (7,5 mmol/l) in pazienti con malattia maligna attiva che non ricevono né chemioterapia né radioterapia. I farmaci stimolanti l'eritropoiesi sono controindicati in questo gruppo di pazienti;
- aumento del 9% del rischio di progressione della malattia o di morte nel gruppo di pazienti che ricevevano epoetina alfa e trattamento standard, e un aumento del 15% del rischio, statisticamente non escludibile, nelle pazienti con cancro al seno metastatico in chemioterapia, quando si mirava a un livello di emoglobina di 10–12 g/dl (6,2–7,5 mmol/l).
Alla luce di quanto sopra, in alcune situazioni cliniche può essere preferibile la trasfusione di sangue per il trattamento dell'anemia nei pazienti oncologici. La decisione sull'uso di eritropoietine ricombinanti deve basarsi su una valutazione del rapporto rischio-beneficio per il singolo paziente, tenendo conto del contesto clinico specifico. I fattori da considerare in tale valutazione devono includere il tipo e lo stadio del tumore; il grado di anemia; la sopravvivenza prevista; le condizioni di trattamento del paziente e le preferenze del paziente stesso.
Nei pazienti con neoplasie in chemioterapia, di solito si osserva un ritardo di 2–3 settimane tra la somministrazione dell'eritropoietina e l'insorgenza delle cellule ematiche indotte. Questa caratteristica deve essere considerata nella valutazione dell'idoneità al trattamento (specialmente nei pazienti con necessità di trasfusioni).
Pazienti sottoposti a intervento chirurgico e partecipanti a un programma di raccolta del sangue autologo.
Devono essere seguite tutte le precauzioni specifiche relative al programma di raccolta del sangue autologo, in particolare le procedure per il ripristino del volume ematico circolante.
Pazienti in previsione di un intervento ortopedico elettivo esteso.
Si devono sempre seguire le pratiche corrette di ematotrasfusione nel periodo pre- e post-operatorio. I pazienti in previsione di un intervento ortopedico elettivo esteso devono ricevere una profilassi antitrombotica adeguata, poiché dopo gli interventi chirurgici in questi pazienti possono verificarsi complicanze trombotiche e vascolari, specialmente in presenza di malattie cardiovascolari concomitanti. Particolare cautela deve essere esercitata nel trattamento di pazienti predisposti allo sviluppo di trombosi venosa profonda. Inoltre, nei pazienti con livello iniziale di emoglobina > 13 g/dl, il rischio di complicanze trombotiche o vascolari post-operatorie associate al trattamento con epoetina alfa è significativamente più elevato. Pertanto, l'uso di epoetina alfa in pazienti con livello iniziale di emoglobina > 13 g/dl non è raccomandato.
Dieta
Con l'aumento dell'ematocrito, nei pazienti aumenta l'appetito, con conseguente aumento dell'assunzione di cibo. In tali casi è necessario adottare misure per prevenire l'aumento del contenuto di potassio nell'organismo.
Proprietà cancerogene e mutagene
Le proprietà cancerogene di Hemax non sono state studiate. L'epoetina non induce mutazioni genetiche nei batteri né aberrazioni cromosomiche nelle cellule di mammiferi.
Effetto sulla fertilità
In ratti gravidi si è osservata una tendenza a un lieve aumento della frequenza di morte embrionale dopo somministrazione endovenosa di epoetina alla dose di 100–500 UI/kg.
Diminuzione dell'efficacia del medicinale o mancanza di risposta
Se nei pazienti in trattamento con epoetina a dosi di mantenimento si osserva una riduzione della risposta al medicinale o una completa mancanza di risposta, è necessario escludere le seguenti cause:
- carenza di ferro
- infezione, infiammazione, tumori
- perdita di sangue occulto
- malattie ematologiche (talassemia, displasia mieloide, ecc.)
- emolisi
- intossicazione da alluminio
- carenza di vitamina B12 o acido folico
- fibrosi cistica
- eritroplasia vera.
Cloruro di sodio
Questo medicinale contiene meno di 1 mmol (23 mg) di sodio per dose, cioè è praticamente privo di sodio.
Uso durante la gravidanza o l'allattamento.
Gravidanza
Non sono stati condotti studi adeguati sull'uso di Hemax in donne in gravidanza. Gli studi sugli animali hanno mostrato tossicità riproduttiva. Pertanto, questo medicinale può essere somministrato in gravidanza solo se il beneficio potenziale giustifica il rischio potenziale per il feto. L'uso di epoetina alfa in donne in gravidanza che partecipano a un programma di raccolta del sangue autologo non è raccomandato.
Negli studi su ratti gravidi si è osservato un lieve aumento della frequenza di morte embrionale, mentre in conigli gravidi non sono stati osservati effetti avversi alla dose di 500 UI/kg.
Allattamento al seno.
L'eritropoietina umana è normalmente presente nel latte materno, sebbene il ruolo dell'eritropoietina assunta con il latte materno rimanga sconosciuto. Non è noto se l'epoetina alfa esogena venga escreta nel latte materno. L'epoetina alfa deve essere usata con cautela nelle donne che allattano. La decisione di continuare o interrompere l'allattamento o di continuare o interrompere il trattamento con epoetina alfa deve essere presa considerando il beneficio dell'allattamento al seno per il bambino e il beneficio del trattamento con epoetina alfa per la donna.
L'uso di epoetina alfa in pazienti che partecipano a un programma di raccolta del sangue autologo durante l'allattamento non è raccomandato.
Fertilità.
Non sono stati condotti studi sull'effetto dell'epoetina alfa sulla fertilità negli uomini o nelle donne.
Capacità di influenzare la velocità di reazione nella guida di autoveicoli o nell'uso di macchinari.
Non sono stati condotti studi specifici a questo riguardo, ma, considerando che durante l'uso del medicinale Hemax possono verificarsi reazioni avverse come cefalea, affaticamento e altre, si raccomanda di astenersi dalla guida di autoveicoli e dall'uso di macchinari da parte di persone che assumono questo medicinale.
Modalità e dosi di somministrazione.
Tutte le altre cause di anemia (carenza di ferro, acido folico, vitamina B12, intossicazione da alluminio, infezione o infiammazione, perdita di sangue, emolisi o fibrosi del midollo osseo di qualsiasi eziologia) devono essere identificate e trattate prima di iniziare la terapia con epoetina alfa e, se necessario, aumentare il dosaggio. Per ottenere una risposta ottimale al trattamento con epoetina alfa, è necessario garantire un adeguato apporto di ferro nell'organismo e, se necessario, prescrivere in aggiunta farmaci a base di ferro (vedere il paragrafo «Proprietà farmacologiche»).
Trattamento dell'anemia sintomatica in pazienti adulti con insufficienza renale cronica.
I sintomi e le complicanze dell'anemia possono variare in base all'età, al sesso e alle patologie concomitanti; è necessaria una valutazione individuale del decorso della malattia e dello stato del paziente da parte del medico.
Il livello raccomandato di emoglobina desiderabile è compreso tra 10 g/dl e 12 g/dl (6,2–7,5 mmol/l). Hemax viene utilizzato per aumentare il livello di emoglobina fino a un massimo di 12 g/dl (7,5 mmol/l). È necessario evitare un aumento del livello di emoglobina superiore a 2 g/dl (1,25 mmol/l) entro 4 settimane. In tal caso, la dose deve essere ridotta come indicato più avanti.
A causa delle caratteristiche individuali, in alcuni pazienti possono talvolta verificarsi livelli di emoglobina superiori o inferiori rispetto all'obiettivo desiderato. Il livello di emoglobina deve essere controllato mediante aggiustamento della dose, tenendo conto del livello raccomandato compreso tra 10 g/dl (6,2 mmol/l) e 12 g/dl (7,5 mmol/l).
È necessario evitare un livello costante di emoglobina superiore a 12 g/dl (7,5 mmol/l). Se la concentrazione di emoglobina aumenta di oltre 2 g/dl (1,25 mmol/l) al mese o se il livello costante di emoglobina supera 12 g/dl (7,5 mmol/l), la dose di Hemax deve essere ridotta del 25%. Se il livello di emoglobina supera 13 g/dl (8,1 mmol/l), il trattamento deve essere sospeso fino a quando il livello di emoglobina non scende a 12 g/dl (7,5 mmol/l), dopodiché il trattamento con epoetina alfa deve essere ripreso con una dose inferiore del 25% rispetto alla dose precedente.
Lo stato dei pazienti deve essere attentamente monitorato per assicurarsi che venga utilizzata la dose minima efficace di Hemax necessaria per controllare l'anemia e i suoi sintomi, mantenendo il livello di emoglobina non superiore a 12 g/dl (7,5 mmol/l).
È necessario aumentare con cautela le dosi degli agenti stimolanti l'eritropoiesi nei pazienti con insufficienza renale cronica. Nei pazienti con scarsa risposta al trattamento con tali agenti, devono essere considerate altre possibili cause di risposta inadeguata (vedere il paragrafo «Proprietà farmacologiche»).
Il trattamento con Hemax è suddiviso in due fasi: fase correttiva e fase di mantenimento.
Pazienti adulti in emodialisi.
Nei pazienti in emodialisi, si preferisce la somministrazione endovenosa.
Fase correttiva.
Dose iniziale: 50 UI/kg 3 volte alla settimana.
Se necessario, la dose può essere aumentata o ridotta di 25 UI/kg (3 volte alla settimana) fino al raggiungimento del livello desiderato di emoglobina compreso tra 10–12 g/dl (6,2–7,5 mmol/l). Questa regolazione della dose deve essere effettuata gradualmente, non più di una volta ogni 4 settimane.
Fase di mantenimento.
La dose settimanale raccomandata è compresa tra 75 e 300 UI/kg.
Per mantenere il livello desiderato di emoglobina compreso tra 10 e 12 g/dl (6,2–7,5 mmol/l), la dose deve essere adeguatamente regolata.
Ai pazienti con livelli iniziali molto bassi di emoglobina (< 6 g/dl, o < 3,75 mmol/l) può essere necessaria una dose di mantenimento più elevata rispetto ai pazienti con anemia meno grave all'inizio del trattamento (emoglobina > 8 g/dl, o > 5 mmol/l).
Pazienti adulti con insufficienza renale pre-dialitica.
Nei pazienti con insufficienza renale pre-dialitica, in assenza di un catetere endovenoso posizionato, il farmaco può essere somministrato per via sottocutanea.
Fase correttiva.
Dose iniziale: 50 UI/kg 3 volte alla settimana, con successivo aumento, se necessario, di 25 UI/kg (3 volte alla settimana), con intervalli tra gli aumenti di dose di almeno 4 settimane, fino al raggiungimento di un livello di emoglobina compreso tra 10–12 g/dl (6,2–7,5 mmol/l).
Fase di mantenimento.
Durante la fase di mantenimento, Hemax può essere somministrato 3 volte alla settimana oppure, in caso di somministrazione sottocutanea, 1 volta alla settimana o 1 volta ogni 2 settimane.
Le dosi e gli intervalli tra le somministrazioni devono essere regolati per mantenere il livello desiderato di emoglobina compreso tra 10 e 12 g/dl (6,2–7,5 mmol/l). L'allungamento degli intervalli tra le somministrazioni può richiedere un aumento della dose.
La dose massima non deve superare 150 UI/kg 3 volte alla settimana, 240 UI/kg (massimo fino a 20000 UI) 1 volta alla settimana o 480 UI/kg (massimo fino a 40000 UI) 1 volta ogni 2 settimane.
Pazienti adulti in dialisi peritoneale.
In assenza di un catetere endovenoso posizionato, Hemax può essere somministrato per via sottocutanea.
Fase correttiva.
Dose iniziale: 50 UI/kg 2 volte alla settimana.
Fase di mantenimento.
La dose raccomandata di mantenimento è compresa tra 25 e 50 UI/kg 2 volte alla settimana, mediante somministrazione di due iniezioni equivalenti.
Per mantenere il livello desiderato di emoglobina compreso tra 10 e 12 g/dl (6,2–7,5 mmol/l), la dose deve essere adeguatamente regolata.
Trattamento dell'anemia indotta da chemioterapia in pazienti adulti.
I sintomi e le complicanze dell'anemia possono variare in base all'età, al sesso e alle patologie concomitanti; è necessaria una valutazione individuale del decorso della malattia e dello stato del paziente da parte del medico.
Hemax deve essere somministrato ai pazienti con anemia (livello di emoglobina ≤ 10 g/dl (6,2 mmol/l)). Dose iniziale: 150 UI/kg per via sottocutanea 3 volte alla settimana.
In alternativa, Hemax può essere somministrato alla dose di 450 UI/kg per via sottocutanea 1 volta alla settimana.
Per mantenere il livello desiderato di emoglobina compreso tra 10 e 12 g/dl (6,2–7,5 mmol/l), la dose deve essere adeguatamente regolata.
A causa delle caratteristiche individuali, in alcuni pazienti possono talvolta verificarsi livelli di emoglobina superiori o inferiori rispetto all'obiettivo desiderato. Il livello di emoglobina deve essere controllato mediante aggiustamento della dose, tenendo conto del livello raccomandato compreso tra 10 g/dl (6,2 mmol/l) e 12 g/dl (7,5 mmol/l). È necessario evitare un livello costante di emoglobina superiore a 12 g/dl (7,5 mmol/l).
Se dopo 4 settimane di trattamento il livello di emoglobina aumenta di almeno 1 g/dl (0,6 mmol/l) o il numero di reticolociti aumenta di ≥ 40000 cellule/µl, il trattamento con Hemax deve proseguire alla dose di 150 UI/kg 3 volte alla settimana oppure 450 UI/kg 1 volta alla settimana.
Se dopo 4 settimane di trattamento con la dose iniziale il livello di emoglobina aumenta di meno di 1 g/dl (0,62 mmol/l) o il livello di reticolociti aumenta di meno di 40000 cellule/µl, la dose deve essere aumentata a 300 UI/kg 3 volte alla settimana.
Se dopo ulteriori 4 settimane di trattamento alla dose di 300 UI/kg 3 volte alla settimana il livello di emoglobina aumenta di 1 g/dl (0,62 mmol/l) o più oppure il numero di reticolociti aumenta di 40000 cellule/µl o più, il trattamento con Hemax deve proseguire alla dose di 300 UI/kg 3 volte alla settimana.
Tuttavia, se il livello di emoglobina aumenta di meno di 1 g/dl (meno di 0,62 mmol/l) o il livello di reticolociti aumenta di meno di 40000 cellule/µl rispetto al livello iniziale, la risposta clinica è considerata incerta e il trattamento deve essere interrotto.
Regolazione della dose per mantenere il livello obiettivo di emoglobina compreso tra 10–12 g/dl.
Se il livello di emoglobina aumenta di oltre 2 g/dl (1,25 mmol/l) in 1 mese oppure se il livello di emoglobina supera 12 g/dl (7,5 mmol/l), la dose di Hemax deve essere ridotta del 25–50%. Se il livello di emoglobina supera 13 g/dl (8,1 mmol/l), la terapia deve essere temporaneamente sospesa fino a quando il livello scende al di sotto di 12 g/dl (7,5 mmol/l), dopodiché la terapia deve essere ripresa con una dose inferiore del 25% rispetto alla dose precedente.
Schema del regime di dosaggio raccomandato:
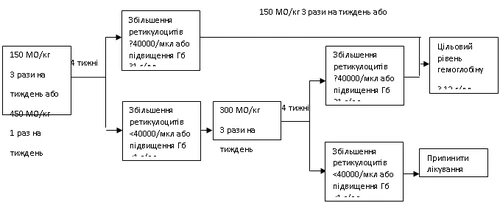
Lo stato dei pazienti deve essere attentamente monitorato per assicurarsi che venga utilizzata la dose minima approvata di Hemax necessaria per controllare i sintomi dell'anemia.
Il trattamento con Hemax deve proseguire per un mese dopo la sospensione della chemioterapia.
Pazienti adulti che partecipano a un programma di prelievo autologo del sangue prima di interventi chirurgici.
Ai pazienti con anemia di grado moderato (ematocrito 33–39%) che necessitano di ≥ 4 unità di sangue, deve essere somministrato Hemax alla dose di 600 UI/kg per via endovenosa 2 volte alla settimana per 3 settimane prima dell'intervento chirurgico. Hemax viene somministrato al termine di ogni procedura di prelievo del sangue.
Pazienti adulti prima di un intervento ortopedico chirurgico maggiore programmato.
Dose raccomandata: 600 UI/kg per via sottocutanea 1 volta alla settimana per 3 settimane precedenti l'intervento (21°, 14° e 7° giorno prima dell'intervento) e nel giorno dell'intervento.
Se per motivi clinici è necessario ridurre il periodo preoperatorio a meno di 3 settimane, Hemax deve essere somministrato giornalmente alla dose di 300 UI/kg per via sottocutanea per 10 giorni consecutivi prima dell'intervento, nel giorno dell'intervento e per 4 giorni successivi. Se nel periodo preoperatorio il livello di emoglobina raggiunge 15 g/dl o superiore, l'uso di Hemax deve essere completamente interrotto e le dosi successive non devono essere somministrate.
Pazienti adulti con SDM a rischio basso o intermedio-1
Hemax deve essere somministrato ai pazienti con anemia sintomatica (livello di emoglobina ≤ 10 g/dl (6,2 mmol/l)).
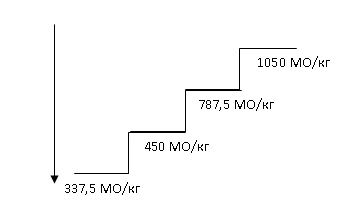
La dose iniziale raccomandata di Hemax è 450 UI/kg (dose totale massima 40000 UI) per via sottocutanea 1 volta alla settimana, con un intervallo minimo di 5 giorni tra le somministrazioni.
La dose deve essere adeguatamente regolata per mantenere il livello desiderato di emoglobina compreso tra 10 e 12 g/dl (6,2–7,5 mmol/l). La risposta al trattamento con Hemax deve essere valutata tra le 8 e le 12 settimane dall'inizio del trattamento. Gli aumenti e le riduzioni della dose devono essere effettuati gradualmente (vedere il diagramma seguente). È necessario evitare un livello di emoglobina superiore a 12 g/dl (7,5 mmol/l).
Aumento della dose. La dose non deve essere aumentata di oltre 1050 UI/kg (dose totale 80000 UI) alla settimana. Se si perde la risposta al trattamento o se la concentrazione di emoglobina diminuisce di ≥ 1 g/dl dopo una riduzione della dose, la dose deve essere aumentata di un livello. Tra gli aumenti della dose devono trascorrere almeno 4 settimane.
Mantenimento e riduzione della dose. Il trattamento con Hemax deve essere sospeso se la concentrazione di emoglobina supera 12 g/dl (7,5 mmol/l). Non appena il livello di emoglobina scende al di sotto di 11 g/dl, la somministrazione deve essere ripresa alla stessa dose o a un livello inferiore, a seconda della decisione del medico. Deve essere presa in considerazione una riduzione della dose se si osserva un rapido aumento del livello di emoglobina (> 2 g/dl in 4 settimane).
 |
|||||
  |
|||||
I sintomi e le complicanze dell'anemia possono variare in base all'età, al sesso e alle malattie concomitanti; è pertanto necessaria una valutazione individuale del decorso della malattia e dello stato del paziente.
Età pediatrica.
Trattamento dell'anemia sintomatica nei bambini con insufficienza renale cronica in emodialisi.
I sintomi e le complicanze dell'anemia possono variare in base all'età, al sesso e alle malattie concomitanti; è pertanto necessaria una valutazione individuale del decorso della malattia e dello stato del paziente.
Nei bambini, il livello raccomandato di emoglobina è compreso tra 9,5 g/dl e 11 g/dl (5,9–6,8 mmol/l). Hemax deve essere utilizzato per aumentare il livello di emoglobina fino a un massimo di 11 g/dl (6,8 mmol/l). È necessario evitare un aumento del livello di emoglobina superiore a 2 g/dl (1,25 mmol/l) entro 4 settimane.
In tal caso, la dose deve essere adeguata in modo appropriato.
Lo stato del paziente deve essere attentamente monitorato al fine di garantire che venga utilizzata la dose più bassa approvata di Hemax necessaria per controllare i sintomi dell'anemia.
Il trattamento è suddiviso in due fasi: fase correttiva e fase di mantenimento.
In presenza di un catetere venoso centrale già posizionato, si preferisce la somministrazione endovenosa.
Fase correttiva.
Dose iniziale: 50 UI/kg endovena 3 volte alla settimana.
Se necessario, la dose può essere ridotta o aumentata di 25 UI/kg (3 volte alla settimana) fino al raggiungimento del livello desiderato di emoglobina compreso tra 9,5 g/dl e 11 g/dl (5,9–6,8 mmol/l). Questa regolazione della dose deve avvenire in modo graduale, non più di una volta ogni 4 settimane.
Fase di mantenimento.
La dose deve essere adeguata in modo appropriato al fine di mantenere un livello ottimale di emoglobina compreso tra 9,5 g/dl e 11 g/dl (5,9–6,8 mmol/l).
In generale, i bambini con un peso corporeo inferiore a 30 kg richiedono una dose di mantenimento più elevata rispetto agli adulti e ai bambini con peso corporeo superiore a 30 kg. I pazienti pediatrici con un livello iniziale di emoglobina molto basso (< 6,8 g/dl, o < 4,25 mmol/l) possono richiedere dosi di mantenimento più elevate rispetto ai pazienti con livello iniziale di emoglobina più alto (> 6,8 g/dl, o > 4,25 mmol/l).
Trattamento dell'anemia nei bambini con insufficienza renale cronica nel periodo pre-dialitico o in dialisi peritoneale.
La sicurezza e l'efficacia dell'uso di Hemax nei bambini con insufficienza renale cronica e anemia non ancora in dialisi o in dialisi peritoneale non sono state stabilite. Attualmente non è possibile fornire raccomandazioni sul dosaggio per questo gruppo di pazienti.
Trattamento dei bambini con anemia indotta da chemioterapia
La sicurezza e l'efficacia dell'uso di Hemax nei bambini con anemia indotta da chemioterapia non sono state stabilite.
Trattamento dei bambini che partecipano a programmi di prelievo di sangue autologo prima di interventi chirurgici.
La sicurezza e l'efficacia dell'uso di Hemax nei bambini che partecipano a programmi di prelievo di sangue autologo prima di interventi chirurgici maggiori non sono state stabilite.
Trattamento dei bambini prima di un intervento ortopedico programmato
La sicurezza e l'efficacia dell'uso di Hemax nei bambini sottoposti a intervento ortopedico programmato non sono state stabilite.
Modalità di somministrazione
L'epoetina alfa può essere somministrata per via sottocutanea o endovenosa.
Modalità di preparazione della soluzione per iniezione: ad ogni flaconcino con il liofilizzato aggiungere la quantità di acqua per preparazioni iniettabili indicata nella tabella seguente.
| Ingrediente attivo |
1000 UI |
2000 UI |
3000 UI |
4000 UI |
10 000 UI |
20 000 UI |
40 000 UI |
| Acqua per preparazioni iniettabili |
1 ml |
2 ml |
2 ml |
2 ml |
1 ml |
1 ml |
1 ml |
Come per tutti i medicinali parenterali, il medicinale epoetina alfa deve essere ispezionato prima dell'uso per verificare l'assenza di particelle visibili e di alterazioni del colore della soluzione.
Somministrazione endovenosa.
Ai pazienti adulti che partecipano a programmi di predeposito del sangue autologo prima di interventi chirurgici, Hemax viene somministrato per via endovenosa. Nel trattamento dell'anemia sintomatica negli adulti e nei bambini con insufficienza renale cronica, in presenza di accesso endovenoso (pazienti in emodialisi), si preferisce la somministrazione endovenosa.
Hemax viene somministrato mediante iniezione endovenosa lenta della durata di 1-5 minuti, a seconda della dose del medicinale. Nei pazienti sottoposti a emodialisi, l'iniezione in bolo può essere effettuata direttamente durante la procedura attraverso un'apposita via venosa nel circuito di dialisi. Il medicinale può anche essere somministrato al termine della procedura di emodialisi attraverso la fistola del catetere, seguito dall'infusione di 10 ml di soluzione fisiologica di sodio cloruro per il lavaggio del circuito e un adeguato distribuzione del medicinale nella circolazione sistemica.
La somministrazione lenta è consigliata nei pazienti che manifestano sintomi simil-influenzali durante il trattamento (vedi sezione «Effetti indesiderati»).
Hemax non deve essere somministrato mediante infusione endovenosa né miscelato con altri medicinali.
Somministrazione sottocutanea.
Ai pazienti adulti con anemia indotta da chemioterapia, ai pazienti in previsione di un intervento chirurgico ortopedico maggiore programmato e ai pazienti adulti con sindrome mielodisplastica a basso rischio o rischio intermedio-1, Hemax viene somministrato per via sottocutanea.
Nel trattamento dell'anemia sintomatica negli adulti con insufficienza renale cronica nel periodo pre-dialitico o in dialisi peritoneale, in assenza di catetere endovenoso impiantato, Hemax può essere somministrato per via sottocutanea.
Il volume massimo di somministrazione sottocutanea del medicinale in un singolo sito è di 1 ml. Se necessario, per dosi superiori, la somministrazione sottocutanea deve essere effettuata in più siti.
Il medicinale deve essere somministrato per via sottocutanea negli arti o nella parete anteriore dell'addome.
Se il medico ritiene che il paziente o il caregiver possa somministrare Hemax sottocutaneamente in modo sicuro ed efficace, è necessario fornire istruzioni adeguate sul dosaggio e sull'uso corretto.
Bambini.
L'epoetina alfa è indicata nel trattamento dell'anemia associata all'insufficienza renale cronica nei bambini di età compresa tra 1 anno e 18 anni sottoposti a dialisi. La sicurezza e l'efficacia del medicinale nei bambini di età inferiore a 1 mese non sono state stabilite.
Sovradosaggio.
Il medicinale ha un ampio intervallo terapeutico. In caso di sovradosaggio di epoetina alfa si possono verificare effetti che riflettono il massimo grado di espressione dell'azione farmacologica dell'ormone, ossia policitemia e sintomi correlati come cefalea, vertigini, sonnolenza, ecc. In caso di livelli emoglobinici eccessivamente elevati, può essere indicata una flebotomia. Se necessario, si applica un trattamento sintomatico.
In caso di sovradosaggio del medicinale, è necessario rivolgersi al più vicino centro medico o centro antiveleni.
Effetti indesiderati.
L'effetto indesiderato più comune durante il trattamento con epoetina alfa è l'aumento dose-dipendente della pressione arteriosa o il peggioramento di una ipertensione preesistente. Il controllo della pressione arteriosa deve essere effettuato fin dall'inizio del trattamento. Altri effetti indesiderati comuni osservati negli studi clinici con epoetina alfa includono trombosi venosa profonda, embolia polmonare, convulsioni, diarrea, nausea, cefalea, stato simil-influenzale, piressia, eruzioni cutanee e vomito.
All'inizio del trattamento possono manifestarsi sintomi simili a quelli del raffreddore, come cefalea, dolore muscolare e articolare e brividi. La frequenza può variare a seconda dell'indicazione.
Durante gli studi sull'uso del medicinale con intervalli tra le dosi prolungati in pazienti adulti con insufficienza renale nel periodo pre-dialitico, è stato osservato un peggioramento della pervietà delle vie respiratorie, comprese le vie respiratorie superiori, congestione nasale e nasofaringite.
Nei pazienti trattati con agenti stimolanti l'eritropoiesi è stata osservata una maggiore frequenza di complicanze trombovascolari (vedi sezione «Informazioni importanti sull'uso del medicinale»).
Albumina (umana)
Hemax contiene albumina ottenuta dal sangue umano. Il rischio di trasmissione di infezioni virali è estremamente basso grazie alla tecnologia impiegata nella produzione di questo componente. Analogamente, il rischio estremamente improbabile riguarda anche la possibile trasmissione dell'agente causale della malattia di Creutzfeldt-Jakob. Non sono noti casi di trasmissione di agenti infettivi virali tramite albumina.
Frequenza degli effetti indesiderati: molto frequente (≥1/10); frequente (≥1/100 fino a <1/10); non frequente (≥1/1000 fino a <1/100); raro (≥1/10000 fino a <1/1000); molto raro (<1/10000); frequenza non nota (non può essere stabilita con i dati disponibili).
Ematologici e del sistema linfatico.
Raro – trombocitemia, aplasia eritroide vera.
Sistema immunitario.
Non frequente – reazioni di ipersensibilità.
Raro – reazioni anafilattiche: complicanze potenzialmente gravi associate a disturbi respiratori o a calo della PA; reazioni immunitarie (ha una minima capacità di indurre la formazione di anticorpi).
Sistema nervoso.
Frequente – cefalea.
Non frequente – convulsioni, ictus, emorragia cerebrale.
Frequenza non nota – ictus cerebrovascolare, encefalopatia ipertensiva, attacco ischemico transitorio, vertigini, sonnolenza.
Organi della vista.
Frequenza non nota – trombosi venosa retinica.
Cuore.
Frequenza non nota – infarto del miocardio.
Sistema vascolare.
Frequente – trombosi venose ed arteriose, ipertensione arteriosa.
Frequenza non nota – trombosi venosa profonda (pazienti con insufficienza renale cronica), crisi ipertensiva.
Apparato respiratorio, torace e mediastino.
Frequente – embolia polmonare (pazienti con cancro), tosse.
Non frequente – peggioramento della pervietà delle vie respiratorie.
Frequenza non nota – embolia polmonare (pazienti con insufficienza renale cronica).
Apparato gastrointestinale.
Molto frequente – nausea, diarrea, vomito.
Pelle e tessuto sottocutaneo.
Frequente – eruzioni cutanee, eczema.
Non frequente – orticaria.
Frequenza non nota – angioedema, prurito, edema di Quincke, sindrome di Stevens-Johnson, necrolisi epidermica tossica (che possono essere potenzialmente letali o avere esito fatale).
Apparato muscolo-scheletrico e tessuto connettivo.
Frequente – artralgia, dolore osseo, dolore agli arti, mialgia.
Malattie congenite, ereditarie/genetiche.
Raro – porfiria acuta.
Disturbi generali e condizioni in sede di somministrazione.
Molto frequente – febbre, piressia (pazienti con cancro).
Frequente – stato simil-influenzale, brividi, reazioni in sede di iniezione, edema periferico.
Frequenza non nota – brividi, mancata risposta al trattamento.
Esami di laboratorio.
Raro – presenza di anticorpi contro l'eritropoietina.
Frequenza non nota – iperfosfatemia, aumento della concentrazione di urea, creatinina e acido urico nel plasma sanguigno (nei pazienti con insufficienza renale cronica).
Metabolismo e nutrizione.
Non frequente – iperkaliemia (frequente nei pazienti in emodialisi).
Lesioni, avvelenamenti e complicanze procedurali.
Frequente – trombosi dello shunt, compresi i dispositivi per dialisi (pazienti con insufficienza renale cronica).
Pazienti con insufficienza renale cronica.
Nei pazienti con insufficienza renale cronica, un livello di emoglobina superiore a 12 g/dl può essere associato a un aumentato rischio di complicanze cardiovascolari, compreso l'esito fatale.
Nei pazienti sottoposti a emodialisi, specialmente in caso di predisposizione all'ipotensione o presenza di complicanze a livello della fistola artero-venosa (stenosi, aneurismi, ecc.), sono stati riportati casi di trombosi dello shunt.
Pazienti con neoplasie.
Può verificarsi lo sviluppo di complicanze trombotiche nei pazienti trattati con terapie stimolanti l'eritropoiesi, inclusa l'epoetina alfa.
Pazienti chirurgici adulti.
Non può essere esclusa la possibilità che il trattamento con epoetina alfa in pazienti con livelli stabili di emoglobina >13 g/dl sia associato a un aumentato rischio di complicanze trombotiche/vascolari post-operatorie.
Descrizione di singoli effetti indesiderati.
Trombosi venose ed arteriose, con esito fatale o senza, come trombosi venosa profonda, embolia polmonare, trombosi retinica, trombosi arteriosa (inclusi infarto miocardico e ischemia miocardica), trombosi della retina e trombosi dello shunt (inclusa l'occlusione del sistema di dialisi), trombosi a livello dell'anastomosi artero-venosa. Possono inoltre verificarsi complicanze cerebrovascolari (inclusi ictus ischemico ed emorragie cerebrali) e attacchi ischemici transitori, aneurismi.
Sono state riportate reazioni di ipersensibilità, in particolare con eruzioni cutanee (inclusa orticaria), reazioni anafilattiche e angioedema.
Sono stati riportati casi gravi di effetti indesiderati cutanei correlati al trattamento con epoetine, inclusa la sindrome di Stevens-Johnson e la necrolisi epidermica tossica, che possono essere potenzialmente letali o avere esito fatale (vedi sezione «Informazioni importanti sull'uso del medicinale»).
Sono stati osservati casi di crisi ipertensiva con encefalopatia e convulsioni, che richiedevano immediata valutazione medica e terapia intensiva, in pazienti con pressione arteriosa normale o bassa all'inizio del trattamento. Si raccomanda particolare attenzione alla comparsa di cefalea improvvisa, pulsante e simile all'emicrania, che può rappresentare un segnale di allarme.
Aplasia eritroide vera.
Poiché l'epoetina alfa è un prodotto proteico, in alcuni pazienti può verificarsi la formazione di anticorpi. Casi rari di aplasia eritroide vera, che si manifestano in pazienti con insufficienza renale in trattamento sottocutaneo, sono associati alla presenza di anticorpi neutralizzanti contro l'epoetina alfa. In tali casi, l'uso di qualsiasi medicinale contenente epoetina è controindicato.
Molto raramente (< 10000 casi per paziente-anno) sono stati riportati casi di aplasia eritroide vera mediata da anticorpi (PRCA) in pazienti trattati con prodotti di eritropoietina per mesi o anni.
Pazienti adulti con MDS a rischio basso o intermedio-1.
Durante uno studio clinico, in 4 (4,7%) pazienti si sono verificati eventi trombovascolari (morte improvvisa, ictus ischemico, embolia e flebite). Tutti gli eventi trombovascolari si sono verificati nel gruppo trattato con epoetina alfa e durante le prime 24 settimane di trattamento. Tre casi sono stati confermati, il quarto caso (morte improvvisa) non è stato confermato. Due pazienti presentavano fattori di rischio significativi (fibrillazione atriale, insufficienza cardiaca e tromboflebite).
Bambini con insufficienza renale cronica in emodialisi.
L'esperienza sull'uso di eritropoietina nei bambini con insufficienza renale cronica in emodialisi, durante studi clinici e nel periodo post-marketing, è limitata. Non sono stati osservati effetti indesiderati specifici per l'età pediatrica, né effetti indesiderati non correlati alla patologia esistente.
La segnalazione degli effetti indesiderati dopo l'autorizzazione del medicinale è importante. Permette un monitoraggio continuo del rapporto beneficio/rischio del medicinale. I professionisti sanitari sono invitati a segnalare qualsiasi effetto indesiderato potenziale attraverso il sistema nazionale di segnalazione.
Durata della validità. 2 anni.
Condizioni di conservazione.
Conservare in luogo inaccessibile ai bambini, a temperatura non superiore a 25 °C.
Confezionamento.
1 flaconcino per confezione in cartone.
Categoria di prescrizione. Sotto prescrizione medica.
Produttore. Biosidus S.A. /
Biosidus S.A.
Indirizzo del produttore e sede legale.
Constitucion 4234 (cap C1254ABX), Città di Buenos Aires, Repubblica Argentina /
Constitucion 4234 (zip code C1254ABX), of the City of Buenos Aires, Argentine Republic.