Rixubis
UcraniaContenido
INSTRUCCIONES para uso médico del medicamento RIXUBIS
Composición:
Principio activo: nonacog gamma*;
1 vial contiene:
250 UI** de nonacog gamma, factor de coagulación sanguínea humana recombinante IX (ADNr), correspondiente a una concentración de 50 UI/ml tras la reconstitución del medicamento con 5 ml de disolvente;
500 UI** de nonacog gamma, factor de coagulación sanguínea humana recombinante IX (ADNr), correspondiente a una concentración de 100 UI/ml tras la reconstitución del medicamento con 5 ml de disolvente;
1000 UI** de nonacog gamma, factor de coagulación sanguínea humana recombinante IX (ADNr), correspondiente a una concentración de 200 UI/ml tras la reconstitución del medicamento con 5 ml de disolvente;
2000 UI** de nonacog gamma, factor de coagulación sanguínea humana recombinante IX (ADNr), correspondiente a una concentración de 400 UI/ml tras la reconstitución del medicamento con 5 ml de disolvente;
3000 UI** de nonacog gamma, factor de coagulación sanguínea humana recombinante IX (ADNr), correspondiente a una concentración de 600 UI/ml tras la reconstitución del medicamento con 5 ml de disolvente.
Excipientes: L-histidina, cloruro de sodio, cloruro de calcio, manitol, sacarosa, polisorbato 80.
1 vial de disolvente contiene: agua para inyección – 5 ml.
_________________________________________________________________________
* Nonacog gamma (factor recombinante de coagulación sanguínea IX [ADNr]) es una glucoproteína purificada monocatenaria que contiene 415 aminoácidos. Se produce mediante tecnología de ADN recombinante en una línea celular de ovario de hámster chino.
** La actividad (UI) se determina mediante un ensayo de una sola etapa para factores de coagulación sanguínea según la Farmacopea Europea. La actividad específica del medicamento RIXUBIS es aproximadamente de 200–390 UI/mg de proteína.
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Principales características físico-químicas: polvo blanco o casi blanco; disolvente – solución transparente e incolora.
Grupo farmacoterapéutico. Agentes antihemorrágicos. Factor de coagulación sanguínea IX.
Código ATC B02BD04.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
El medicamento RIXUBIS contiene factor de coagulación de la sangre recombinante IX (nonacog gamma). El factor IX es una glucoproteína de cadena simple con una masa molecular de aproximadamente 68 000 daltons. Es un factor de coagulación dependiente de la vitamina K que se produce en el hígado. El factor IX se activa con la participación del factor XIa en el sistema de coagulación intrínseco y del complejo factor VII/factor tisular en el sistema de coagulación extrínseco. El factor IX activado, junto con el factor VIII activado, activa el factor X. El factor X activado convierte la protrombina en trombina. Posteriormente, la trombina convierte el fibrinógeno en fibrina, formándose así un coágulo.
Farmacodinámica
La hemofilia B es una patología hereditaria ligada al sexo del sistema de coagulación sanguínea, causada por niveles reducidos de factor IX, que provoca hemorragias abundantes en articulaciones, músculos o órganos internos, de forma espontánea o tras traumatismos accidentales o quirúrgicos. La terapia sustitutiva permite aumentar temporalmente los niveles plasmáticos de factor IX, corrigiendo así temporalmente el déficit y reduciendo la predisposición a las hemorragias.
Eficacia clínica y seguridad
Prevención y control de hemorragias en pacientes de 12 años o más que previamente han sido tratados
La eficacia de RIXUBIS se evaluó en la parte no controlada de un estudio combinado de fase 1/3 abierto, en el que 73 pacientes de 12 a 59 años de edad, previamente tratados, recibieron RIXUBIS para profilaxis y/o tratamiento de episodios hemorrágicos "según necesidad". Todos los pacientes tenían hemofilia B grave (nivel de factor IX < 1 %) o moderadamente grave (nivel de factor IX < 2 %). De ellos, 59 pacientes recibieron RIXUBIS para profilaxis. Los datos de 56 pacientes que recibieron RIXUBIS durante al menos 3 meses se incluyeron en el conjunto de datos destinado a evaluar la eficacia de la profilaxis. Otros 14 pacientes recibieron RIXUBIS únicamente para el tratamiento de episodios hemorrágicos. Los pacientes de la cohorte de tratamiento "según necesidad" debían tener al menos 12 episodios hemorrágicos documentados que requirieran tratamiento durante los 12 meses previos a la inclusión en el estudio. La duración media del tratamiento en la cohorte "según necesidad" fue de 3,5 ± 1,00 meses (mediana: 3,4; rango: de 1,2 a 5,1 meses). La frecuencia media anualizada de hemorragias (tasa anualizada de sangrado, ABR) fue de 33,9 ± 17,37, con una mediana de 27,0 y un rango de 12,9 a 73,1.
La mediana de ABR con profilaxis mediante RIXUBIS fue de 2,0 para todas las hemorragias, de 0,0 para hemorragias espontáneas y de 0,0 para hemorragias articulares. En 24 pacientes (42,9 %) no se produjeron hemorragias.
Un total de 249 episodios hemorrágicos fueron tratados con RIXUBIS, de los cuales 197 fueron hemorragias articulares y 52 hemorragias extrarticulares (en tejidos blandos, músculos, cavidades corporales, hemorragia intracraneal y otros). De los 249 episodios, 163 fueron de intensidad moderada, 71 de intensidad leve y 15 graves. El tratamiento se adaptó individualmente según la gravedad, causa y localización de la hemorragia. La mayoría (211 episodios; 84,7 %) de los 249 episodios se trataron con 1-2 infusiones. La eficacia hemostática para detener la hemorragia fue evaluada como excelente o alta en el 96 % de todos los episodios tratados.
Prevención y control de hemorragias en pacientes menores de 12 años que previamente han sido tratados
La eficacia de RIXUBIS se evaluó en un estudio combinado de fase 2/3, en el que 23 pacientes masculinos de 1,8 a 11,8 años (mediana de edad: 7,10 años), previamente tratados, de los cuales 11 tenían menos de 6 años, recibieron RIXUBIS para la profilaxis y control de episodios hemorrágicos. Todos los pacientes tenían hemofilia B grave (nivel de factor IX < 1 %) o moderadamente grave (nivel de factor IX < 2 %). Los 23 pacientes recibieron tratamiento profiláctico con RIXUBIS durante al menos 3 meses y fueron incluidos en el análisis para evaluar la eficacia de la profilaxis.
La mediana de ABR fue de 2,0, de 0,0 para hemorragias espontáneas y de 0,0 para hemorragias articulares.
En nueve pacientes (39,1 %) no se produjeron hemorragias.
Un total de 26 episodios hemorrágicos fueron tratados con RIXUBIS, de los cuales 23 fueron traumáticos, 2 espontáneos y 1 de origen desconocido. De ellos, 19 fueron extrarticulares (en tejidos blandos, músculos, cavidades corporales, hemorragia intracraneal y otros) y 7 articulares, uno de los cuales fue una hemorragia en articulación diana. De los 26 episodios, 15 fueron leves, 9 moderados y 2 graves. El tratamiento se adaptó individualmente según la gravedad, causa y localización de la hemorragia. La mayoría (23; 88,5 %) recibieron tratamiento con 1-2 infusiones. La eficacia hemostática para detener la hemorragia fue evaluada como excelente o alta en el 96,2 % de todos los episodios tratados.
Tratamiento perioperatorio
La seguridad y eficacia de RIXUBIS en el tratamiento perioperatorio se evaluaron en un estudio prospectivo, abierto y no controlado, multicéntrico de fase 3, con hombres con hemofilia B grave o moderada previamente tratados. El análisis de eficacia según protocolo incluyó 37 intervenciones quirúrgicas, incluyendo procedimientos mayores y menores, intervenciones dentales u otras intervenciones quirúrgicas invasivas, realizadas en 27 pacientes de 17 a 57 años. Veinte intervenciones fueron mayores, incluyendo 13 procedimientos ortopédicos y 3 intervenciones dentales quirúrgicas. Diecisiete intervenciones, incluyendo 10 extracciones dentales, se consideraron menores. Los pacientes sometidos a cirugía mayor debían haber sido evaluados farmacocinéticamente (PK). Todos los pacientes recibieron una dosis basada en datos recientes de recuperación incremental de la actividad del factor IX. La dosis inicial recomendada de RIXUBIS debía asegurar niveles de actividad del factor IX del 80-100 % durante cirugías mayores y del 30-60 % durante procedimientos menores. RIXUBIS se administró mediante infusión intravenosa en bolo.
Durante todo el período del estudio se mantuvo el control hemostático.
Farmacocinética
Pacientes de 12 años o más que previamente han sido tratados
Se realizó un estudio farmacocinético aleatorizado, ciego y controlado, cruzado, de RIXUBIS y un medicamento de comparación, con hombres sin hemorragia activa (de 15 años o más) dentro del marco de un estudio combinado central de fase 1/3. Los pacientes recibieron uno u otro medicamento mediante una infusión intravenosa única. La dosis media (± desviación estándar) y mediana de RIXUBIS en el conjunto de datos analizado según protocolo (n = 25) fue de 74,69 ± 2,37 y 74,25 UI/kg, respectivamente, con un rango de 71,27 a 79,38 UI/kg. Los parámetros farmacocinéticos se calcularon a partir de mediciones de la actividad del factor IX en muestras de sangre obtenidas durante un período de hasta 72 horas tras cada infusión.
La evaluación farmacocinética de RIXUBIS se repitió en un estudio abierto no controlado con hombres que participaron en el estudio cruzado inicial de PK y que recibieron profilaxis con RIXUBIS durante 26 ± 1 semanas (media ± desviación estándar), con un período total de tratamiento con RIXUBIS de al menos 30 días. El rango de dosis de RIXUBIS utilizadas en el estudio de PK repetido fue de 64,48 a 79,18 UI/kg (n = 23).
En la Tabla 1 se muestran los parámetros farmacocinéticos en todos los pacientes cuyos datos pudieron evaluarse (análisis según protocolo).
Tabla 1
| Parámetro |
RIXUBIS Estudio cruzado inicial (N = 25) |
RIXUBIS Reevaluación (N = 23) |
| AUC0-72 h (MO • h/dl)a Media ± desviación estándar (DE) Mediana (rango) |
1067,81 ± 238,42 1108,35 (696,07–1571,16) |
1156,15 ± 259,44 1170,26 (753,85–1626,81) |
| Aumento de recuperación del nivel de actividad en Cmax (MO/dl : MO/kg)b Media ± DE Mediana (rango) |
0,87 ± 0,22 0,88 (0,53–1,35) |
0,95 ± 0,25 0,93 (0,52–1,38) |
| Período de semidesintegración (h) Media ± DE Mediana (rango) |
26,70 ± 9,55 24,58 (15,83–52,34) |
25,36 ± 6,86 24,59 (16,24–42,20) |
| Cmax (MO/dl) Media ± DE Mediana (rango) |
66,22 ± 15,80 68,10 (41,70–100,30) |
72,75 ± 19,73 72,40 (38,50–106,30) |
| Tiempo medio de retención en el organismo (h) Media ± DE Mediana (rango) |
30,82 ± 7,26 28,93 (22,25–47,78) |
29,88 ± 4,16 29,04 (21,32–37,52) |
| Vssc (dl/kg) Media ± DE Mediana (rango) |
2,02 ± 0,77 1,72 (1,10–3,94) |
1,79 ± 0,45 1,74 (1,12–2,72) |
| Depuración (dl/kg•h) Media ± DE Mediana (rango) |
0,0644 ± 0,0133 0,0622 (0,0426–0,0912) |
0,0602 ± 0,0146 0,0576 (0,0413–0,0945) |
a Área bajo la curva de concentración en plasma-tiempo de 0 a 72 horas tras la infusión.
b Calculado como el valor (Cmáx del factor IX antes del inicio del tratamiento), dividido por la dosis en unidades UI/kg, donde Cmáx es el valor máximo de actividad del factor IX tras la infusión.
c Volumen de distribución en estado estacionario.
El incremento incremental en la recuperación del nivel de actividad del factor IX a los 30 minutos tras la infusión se determinó en todos los pacientes incluidos en el estudio combinado de Fase 1/3, en el día 1 del tratamiento, en las visitas médicas de las semanas 5, 13 y 26, y en el momento de finalización del estudio o de retirada del mismo, si este no coincidía con la visita de la semana 26. Los datos obtenidos indican que el incremento incremental en la recuperación se mantuvo estable a lo largo del tiempo (ver Tabla 2).
Tabla 2
| Indicador |
Día 1 del tratamiento (N = 73) |
Semana 5 (N = 71) |
Semana 13 (N = 68) |
Semana 26 (N = 55) |
Día de finalización o interrupción del estudio b (N = 23) |
| Aumento de recuperación de actividad a los 30 minutos tras la infusión (MO/dl : MO/kg) a Media ± DE Mediana (rango) |
0,79 ± 0,20 0,78 (0,26–1,35) |
0,83 ± 0,21 0,79 (0,46–1,48) |
0,85 ± 0,25 0,83 (0,14–1,47) |
0,89 ± 0,12 0,88 (0,52–1,29) |
0,87 ± 0,20 0,89 (0,52–1,32) |
a Calculado como el valor de C30 min del factor IX antes del inicio del tratamiento, dividido por la dosis en UOI/kg, donde C30 min es la medición de la actividad del factor IX a los 30 minutos tras la infusión.
b Si no coincide con el momento de la visita en la semana 26.
Población pediátrica (niños previamente tratados, menores de 12 años)
En el marco de un estudio combinado de Fase 2/3, se realizó una evaluación inicial de la farmacocinética de RIXUBIS en 23 pacientes pediátricos de sexo masculino sin hemorragia activa. Para reducir la incomodidad derivada de la frecuente extracción de muestras de sangre en cada paciente individual, los pacientes fueron aleatorizados en dos grupos con distinta secuencia de toma de muestras sanguíneas. La dosis media (± desviación estándar) y la mediana de la dosis de RIXUBIS en la población total analizada (n = 23) fueron 75,50 ± 3,016 y 75,25 UOI/kg, respectivamente, con un rango de 70,0 a 83,6 UOI/kg. Los parámetros farmacocinéticos se calcularon basándose en las mediciones de la actividad del factor IX en muestras de sangre obtenidas durante un período de hasta 72 horas tras la infusión.
En la Tabla 3 se muestran los parámetros farmacocinéticos para todos los pacientes (población total analizada).
Tabla 3
| Parámetro |
Hasta 6 años (N = 11) |
De 6 a 12 años (N = 12) |
Todo el grupo (N = 23) |
| AUCinf (µg • h/dL)ᵃ Media ± EE Mediana (rango) |
723,7 ± 119,00 717,2 (488–947) |
886,0 ± 133,66 863,7 (730–1138) |
808,4 ± 149,14 802,9 (488–1138) |
| Período de semidesintegración (h) Media ± EE Mediana (rango) |
27,67 ± 2,66 27,28 (24,0–32,2) |
23,15 ± 1,58 22,65 (21,8–27,4) |
25,31 ± 3,13 24,48 (21,8–32,2) |
| Tiempo medio de retención en el organismo (h) Media ± EE Mediana (rango) |
30,62 ± 3,27 30,08 (26,2–36,2) |
25,31 ± 1,83 24,74 (23,7–30,3) |
27,85 ± 3,73 26,77 (23,7–36,2) |
| Vssᵇ (dL/kg) Media ± EE Mediana (rango) |
3,22 ± 0,52 3,16 (2,65–4,42) |
2,21 ± 0,32 2,185 (1,70–2,70) |
2,7 ± 0,67 2,69 (1,70–4,42) |
| Depuración (dL/kg • h) Media ± EE Mediana (rango) |
0,1058 ± 0,01650 0,1050 (0,081–0,144) |
0,0874 ± 0,01213 0,0863 (0,069–0,108) |
0,0962 ± 0,01689 0,0935 (0,069–0,144) |
a Área bajo la curva de concentración plasmática-tiempo desde 0 horas hasta infinito.
b Volumen de distribución en estado estacionario.
El incremento en la recuperación del nivel de actividad del factor IX a los 30 minutos tras la infusión se determinó en todos los pacientes incluidos en el estudio combinado de Fase 2/3, en la evaluación de los parámetros farmacocinéticos iniciales (en el día 1 del tratamiento), en las visitas médicas en las semanas 5, 13 y 26, y en el momento de finalización del estudio o de retirada del mismo, si este no coincidía con la visita de la semana 26. Los datos obtenidos indican que el incremento en la recuperación de la actividad se mantiene estable a lo largo del tiempo en todos los grupos de edad pediátricos (ver tablas 4, 5 y 6 a continuación).
Tabla 4
Incremento en la recuperación de la actividad tras la administración de RIXUBIS a los 30 minutos después de la infusión en pacientes pediátricos de ambos grupos de edad
| Aumento de la recuperación de la actividad a los 30 minutos tras la infusión |
FC (1.er día de tratamiento) Todos (N = 22) |
5.ª semana Todos (N = 23) |
13.ª semana Todos (N = 21) |
26.ª semana Todos (N = 21) |
| (MO/dl: MO/kg)a Media ± EE Mediana (rango) |
0,67 ± 0,16 0,69 (0,31–1,00) |
0,68 ± 0,12 0,66 (0,48–0,92) |
0,71 ± 0,13 0,66 (0,51–1,00) |
0,72 ± 0,15 0,734 (0,51–1,01) |
a Calculado como el índice (actividad del factor IX a los 30 minutos antes del inicio del tratamiento) dividido por la dosis en UI/kg, donde C30 min es el índice de medición de la actividad del factor IX a los 30 minutos tras la infusión.
Tabla 5
Incremento progresivo de la recuperación de la actividad tras la administración de RIXUBIS a los 30 minutos de la infusión en pacientes pediátricos menores de 6 años de edad
| Aumento de la recuperación de la actividad a los 30 min después de la infusión |
FC (1.er día de tratamiento) Todos (N = 10) |
5.ª semana, Todos (N = 11) |
13.ª semana, Todos (N = 10) |
26.ª semana, Todos (N = 10) |
| (U/dl: U/kg)a Media ± DE Mediana (rango) |
0,59 ± 0,13 0,59 (0,31–0,75) |
0,63 ± 0,10 0,6 (0,49–0,80) |
0,68 ± 0,12 0,66 (0,51–0,84) |
0,65 ± 0,13 0,61 (0,51–0,84) |
a Calculado como el índice (actividad del factor IX a los 30 minutos antes del inicio del tratamiento) dividido por la dosis en unidades UI/kg, donde C30 min es el valor de medición de la actividad del factor IX a los 30 minutos tras la infusión.
Tabla 6
Aumento progresivo de la recuperación de la actividad tras la administración de RIXUBIS a los 30 minutos de la infusión en pacientes pediátricos de 6 a 12 años de edad
| Aumento de recuperación de actividad a los 30 minutos tras la infusión |
FC (1.er día de tratamiento) Todos (N = 12) |
5.º semana, Todos (N = 12) |
13.ª semana, Todos (N = 11) |
26.ª semana, Todos (N = 11) |
| (MO/dl: MO/kg)a Media ± EE Mediana (rango) |
0,73 ± 0,16 0,71 (0,51–1,00) |
0,73 ± 0,13 0,70 (0,48–0,92) |
0,73 ± 0,14 0,70 (0,54–1,00) |
0,8 ± 0,14 0,78 (0,56–1,01) |
a Calculado como el valor (C30 min del factor IX antes del inicio del tratamiento) dividido por la dosis en UI/kg, donde C30 min es el valor de medición de la actividad del factor IX a los 30 minutos tras la infusión.
Características clínicas.
Indicaciones.
Para el tratamiento y profilaxis de las hemorragias en pacientes con hemofilia B (déficit congénito del factor IX).
El medicamento RIXUBIS está indicado para pacientes de todas las edades.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes mencionados en la sección «Composición».
Reacción alérgica conocida a proteínas de hámster.
Interacción con otros medicamentos y otras formas de interacción.
No se han notificado interacciones de los medicamentos basados en el factor de coagulación sanguínea humana IX (ADNr) con otros medicamentos.
Características de uso.
Seguimiento
Para mejorar el seguimiento de los medicamentos biológicos, se debe indicar claramente el nombre y el número de lote del medicamento utilizado, por ejemplo, en el diario personal del paciente.
Hipersensibilidad
Se han notificado reacciones de hipersensibilidad de naturaleza alérgica con el uso del medicamento RIXUBIS. El medicamento contiene una cantidad residual de proteínas de hámster. Si aparecen síntomas de hipersensibilidad, se debe recomendar a los pacientes o a las personas que los cuidan que interrumpan inmediatamente el uso del medicamento y consulten a su médico. Los pacientes deben estar informados sobre los primeros síntomas de una reacción de hipersensibilidad, incluyendo urticaria, urticaria generalizada, opresión en el pecho, dificultad respiratoria con sibilancias, hipotensión y anafilaxia.
El riesgo es mayor en las primeras etapas del tratamiento con concentrados del factor IX en pacientes que nunca antes han sido tratados, especialmente en aquellos con mutaciones genéticas de alto riesgo. En publicaciones se ha informado sobre una relación entre la aparición de inhibidores del factor IX y reacciones alérgicas, especialmente en pacientes con mutaciones genéticas de alto riesgo. Por esta razón, los pacientes que presenten reacciones alérgicas deben ser evaluados para detectar la presencia de tales inhibidores.
En caso de presentarse shock, se debe proporcionar tratamiento médico estándar contra el shock.
Inhibidores
Después del tratamiento repetido con medicamentos basados en el factor de coagulación sanguínea humana IX (rDNA), los pacientes deben ser examinados para detectar la aparición de anticuerpos neutralizantes (inhibidores), cuyo nivel debe determinarse en unidades de Bethesda (UB) mediante el uso de un ensayo biológico apropiado.
En publicaciones se ha informado sobre una relación entre la aparición de inhibidores del factor IX y reacciones alérgicas. Por esta razón, los pacientes con reacciones alérgicas deben ser evaluados para detectar la presencia de tales inhibidores. Los pacientes en los que aparezcan inhibidores del factor IX tienen un mayor riesgo de desarrollar anafilaxia si se repite el tratamiento con factor IX.
Debido al riesgo de reacciones alérgicas con el uso de concentrados del factor IX, la primera administración de factor IX debe realizarse, bajo criterio del médico tratante, bajo supervisión médica, lo que permita garantizar la prestación de la atención médica adecuada en caso de reacciones alérgicas.
Síndrome nefrótico
Después de estimular la inducción de tolerancia inmunológica en pacientes con hemofilia B que tenían inhibidores del factor IX, se han registrado casos de desarrollo de síndrome nefrótico.
Tromboembolismo
Debido al riesgo potencial de complicaciones trombóticas, se debe realizar un seguimiento clínico para detectar signos tempranos de trombosis y coagulopatía consumptiva mediante el análisis biológico adecuado cuando se administre este medicamento a pacientes con enfermedad hepática, pacientes postoperatorios, recién nacidos o pacientes con riesgo de desarrollar fenómenos trombóticos o síndrome de coagulación intravascular diseminada (CID). En cada uno de estos casos, se deben considerar cuidadosamente los beneficios del tratamiento con RIXUBIS frente al riesgo de presentar estas complicaciones.
Enfermedades cardiovasculares
En pacientes con enfermedades cardiovasculares preexistentes, la terapia de reemplazo con factor IX puede aumentar el riesgo de desarrollar patología cardiovascular.
Complicaciones relacionadas con el uso del catéter
Si es necesario utilizar un dispositivo venoso central (DVC), se debe considerar la posibilidad de complicaciones relacionadas con el uso del DVC, incluyendo infecciones locales, bacteriemia y trombosis en el sitio de cateterización.
Consideraciones relacionadas con los excipientes
Tras la reconstitución, este medicamento contiene menos de 1 mmol (23 mg) de sodio por vial, es decir, prácticamente carece de sodio. Dependiendo del peso corporal del paciente y de la dosis prescrita, los pacientes pueden recibir más de un vial de RIXUBIS, lo cual debe tenerse en cuenta si se sigue una dieta con control del contenido de sodio.
Pacientes de edad avanzada
Los pacientes de 65 años o más no fueron incluidos en los estudios clínicos con RIXUBIS. No se sabe si su respuesta al tratamiento difiere de la de pacientes más jóvenes. Como para todos los demás pacientes, la elección de la dosis para el tratamiento de pacientes de edad avanzada debe hacerse de forma individual.
Niños
Las advertencias y precauciones mencionadas anteriormente se aplican tanto al tratamiento de adultos como de niños.
Uso durante el embarazo o la lactancia.
Embarazo
Los datos sobre el uso del factor IX en mujeres durante el embarazo son inexistentes o limitados. No se han realizado estudios sobre el efecto del factor IX en la función reproductiva en animales.
El factor IX debe usarse durante el embarazo y la lactancia solo si claramente está indicado.
Lactancia
No existen datos sobre la excreción del factor IX o sus metabolitos en la leche materna humana.
Fertilidad
No hay información disponible sobre el efecto del factor IX sobre la fertilidad.
Capacidad para conducir vehículos de motor o manejar maquinaria.
RIXUBIS no afecta la capacidad para conducir vehículos de motor o manejar maquinaria.
Vía de administración y dosis.
El tratamiento debe realizarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Control durante el tratamiento
Durante el tratamiento se recomienda realizar una adecuada determinación de los niveles del factor IX como referencia para la prescripción de la dosis y la frecuencia de las infusiones repetidas. La respuesta al factor IX puede variar entre pacientes individuales, con diferentes valores del período de semivida y del grado de recuperación. A los pacientes con bajo o alto peso corporal puede ser necesaria una corrección de la dosis calculada en base al peso corporal. En caso de intervenciones quirúrgicas graves, es extremadamente importante garantizar un control preciso durante la terapia sustitutiva mediante el análisis de los factores de coagulación sanguínea (actividad del factor IX en plasma sanguíneo).
Para asegurar el logro de los niveles deseados de actividad del factor IX en el plasma sanguíneo, se recomienda realizar un control riguroso mediante un método adecuado para la determinación de la actividad del factor IX y, si es necesario, realizar ajustes correspondientes en la dosis y en la frecuencia de las infusiones repetidas. Al utilizar el método uniestadio de análisis in vitro de los factores de coagulación sanguínea mediante el tiempo de tromboplastina parcial activada (TTPa) para determinar la actividad del factor IX en muestras de sangre de pacientes, los resultados respecto a la actividad del factor IX pueden depender significativamente tanto del tipo de reactivo TTPa como del estándar de comparación utilizados en el análisis. Esto es especialmente importante al cambiar de laboratorio y/o de reactivos utilizados para el análisis.
Dosis
La magnitud de la dosis y la duración del curso de terapia sustitutiva dependen del grado de deficiencia del factor IX, de la localización y gravedad de la hemorragia, así como del estado clínico, edad y parámetros farmacocinéticos del factor IX en el paciente, tales como el incremento de recuperación del nivel de actividad del factor IX y su período de semivida.
La cantidad administrada de unidades del factor IX se expresa en unidades internacionales (UI) de acuerdo con el estándar vigente de la OMS para medicamentos basados en el factor IX. La actividad del factor IX en el plasma sanguíneo se expresa bien en porcentaje (en relación con el valor normal del plasma sanguíneo humano), bien en unidades internacionales (de acuerdo con el estándar internacional para el factor IX en plasma sanguíneo).
Una unidad internacional (UI) de actividad del factor IX equivale a la actividad del factor IX presente en 1 ml de plasma sanguíneo humano normal.
Adultos
Tratamiento "a demanda"
El cálculo de la dosis necesaria del factor IX para pacientes a partir de 12 años de edad se basa en la observación empírica de que la administración de una unidad internacional (UI) de factor IX por kg de peso corporal produce un aumento de la actividad del factor IX en plasma de 0,9 UI/dl (rango de 0,5 a 1,4 UI/dl) o del 0,9 % de la actividad normal.
La dosis necesaria se determina mediante la fórmula indicada a continuación.
| Cantidad necesaria de unidades |
= |
peso corporal (kg) |
× |
aumento deseado del nivel del factor IX (% o U/ml) |
× |
inverso del valor del índice de recuperación de la actividad del factor IX registrado (ml/kg) |
Para un aumento escalonado de la actividad hasta alcanzar un nivel de 0,9 UI/ml por 1 UI/kg, la dosis se calcula según la siguiente fórmula:
| Cantidad necesaria de unidades |
= |
masa corporal (kg) |
× |
aumento deseado del nivel de factor IX (% o U/ml) |
× |
1,1 l/kg |
La magnitud de la dosis y la frecuencia de administración previstas deben estar siempre dirigidas a garantizar la eficacia clínica en cada paciente en particular.
En caso de presentarse los fenómenos hemorrágicos indicados a continuación, los niveles de actividad del factor IX no deben ser inferiores al nivel de actividad indicado en el plasma sanguíneo (en % respecto a la normalidad o en UI/dl) durante el período de tiempo correspondiente. La tabla 7 puede utilizarse para determinar la dosis en episodios de hemorragia y durante intervenciones quirúrgicas.
Tabla 7
| Gravedad del sangrado/ tipo de intervención quirúrgica |
Nivel necesario del factor IX, % o U/dl |
Frecuencia de administración (horas)/ duración del tratamiento (días) |
| Sangrado Hemartrosis precoz, hemorragias musculares o hematomas orales |
20–40 |
Repetir cada 24 horas. Al menos 1 día, hasta la detención del sangrado, evidenciada por la ausencia de dolor, o hasta la cicatrización de la herida. |
| Hemartrosis más pronunciada, hemorragias musculares o hematoma |
30–60 |
Repetir la infusión cada 24 horas durante 3–4 días o más, hasta la desaparición del dolor y la recuperación de la función alterada. |
| Sangrados amenazantes para la vida |
60–100 |
Repetir la infusión cada 8–24 horas hasta que desaparezca el riesgo. |
| Intervenciones quirúrgicas Pequeñas intervenciones quirúrgicas, incluyendo extracciones dentales |
30–60 |
Cada 24 horas, al menos 1 día, hasta la cicatrización de la herida. |
| Grandes intervenciones quirúrgicas |
80–100 (antes y después de la cirugía) |
Repetir la infusión cada 8–24 horas hasta una adecuada cicatrización de la herida, tras lo cual continuar el tratamiento al menos otros 7 días para mantener la actividad del factor IX entre el 30 % y el 60 % (U/dl). |
Un control riguroso del tratamiento sustitutivo es especialmente importante en caso de cirugía mayor o en caso de hemorragia potencialmente mortal.
Prevención
Para la prevención a largo plazo de hemorragias en pacientes de 12 años de edad o más con hemofilia B grave, generalmente se administran dosis de 40 a 60 UI de factor IX por kg de peso corporal cada 3-4 días. En algunos casos, dependiendo de la farmacocinética, la edad, el fenotipo hemorrágico y el grado de actividad física del paciente, puede ser necesario acortar los intervalos entre las administraciones o aumentar las dosis del medicamento.
Infusión continua
No administrar el medicamento RIXUBIS mediante infusión continua.
Vía de administración
Administración intravenosa.
Si el medicamento lo administra el propio paciente o una persona encargada de su cuidado, debe recibir una formación adecuada para realizar este procedimiento.
El medicamento RIXUBIS debe administrarse a una velocidad que resulte cómoda para el paciente, con un máximo de hasta 10 ml/min.
Después de la reconstitución, la solución del medicamento es transparente, incolora, sin inclusiones mecánicas extrañas y con un pH de 6,8–7,2. El valor de la osmolaridad es superior a 240 mOsmol/kg.
Para la administración de este medicamento solo deben utilizarse jeringas de plástico con cono de acoplamiento Luer.
El medicamento RIXUBIS se administra por vía intravenosa tras la reconstitución del polvo con el disolvente suministrado.
- Para la preparación de la solución del medicamento, utilice exclusivamente el disolvente y el dispositivo de reconstitución (BAXJECT II) suministrados en el envase.
- Para la administración del medicamento debe utilizarse una jeringa con cono de acoplamiento Luer.
- No utilice el dispositivo BAXJECT II si está dañado, si el sistema estéril de protección o el envase están dañados, o si existen signos de deterioro.
Reconstitución
Aplique técnicas asépticas.
- Si el medicamento se ha almacenado en nevera, retire los frascos con el polvo de RIXUBIS y el disolvente del refrigerador y déjelos alcanzar la temperatura ambiente (entre 15 °C y 30 °C).
- Lávese cuidadosamente las manos con agua tibia y jabón.
- Retire las tapas de los frascos del polvo y del disolvente.
- Limpie los tapones con torundas de alcohol. Coloque los frascos sobre una superficie plana y limpia.
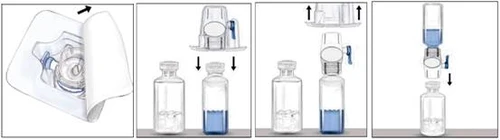
- Abra el envase del dispositivo BAXJECT II retirando la película de papel, sin tocar la superficie interna del dispositivo (figura a). No retire el dispositivo del envase.
- Invierta el envase y coloque la punta plástica transparente sobre el tapón del frasco del disolvente. Sujete el envase por los bordes y retire el envase del dispositivo BAXJECT II (figura b). No retire la tapa azul del dispositivo BAXJECT II.
- Una vez que el dispositivo BAXJECT II esté conectado al frasco del disolvente, invierta el conjunto de modo que el frasco del disolvente quede por encima del dispositivo. Coloque la punta plástica blanca sobre el tapón del frasco del medicamento RIXUBIS. Por efecto del vacío, el disolvente pasará al frasco que contiene el medicamento RIXUBIS (figura c).
- Agite suavemente hasta que el polvo se disuelva completamente. El medicamento se disuelve rápidamente (en aproximadamente 2 minutos). Asegúrese de que RIXUBIS se haya disuelto completamente, ya que, de lo contrario, no todo el medicamento reconstituido pasará a través del filtro del dispositivo. Antes de su uso, los medicamentos reconstituidos deben examinarse visualmente para detectar partículas sólidas y decoloración. La solución debe ser transparente o ligeramente opalescente. No utilice soluciones turbias ni que contengan sedimento.
| Figura a |
Figura b |
Figura c |
|
|
||
No enfríe el medicamento después de la reconstitución. Utilícelo inmediatamente después de su preparación.
Uso
Aplique técnicas asépticas.
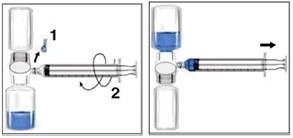
- Retire la tapa azul del dispositivo BAXJECT II. Tenga cuidado de no aspirar aire en la jeringa. Conecte la jeringa al dispositivo BAXJECT II (figura d).
- Invierta el sistema (el frasco con la solución reconstituida debe quedar arriba). Llene la jeringa con la solución reconstituida, tirando lentamente del émbolo hacia atrás (figura e).
- Desconecte la jeringa.
- Conecte una aguja-borla a la jeringa. Administre la solución por vía intravenosa. La solución debe administrarse lentamente, a una velocidad cómoda para el paciente, no más rápida de 10 ml por minuto.
| Figura d |
Figura e |
|
|
|
Siempre que utilice el medicamento RIXUBIS, anote su nombre y número de lote (por ejemplo, en su diario personal) para conservar la información sobre los medicamentos que ha utilizado.
Cualquier resto de medicamento no utilizado o sus desechos deben eliminarse de acuerdo con las normas locales.
La estabilidad química y física de la preparación diluida y lista para su uso es de 3 horas a una temperatura no superior a 30 °C. Desde el punto de vista microbiológico, aunque el procedimiento de dilución excluye la contaminación microbiana, el medicamento debe utilizarse inmediatamente. Si no se utiliza inmediatamente, el período y las condiciones de almacenamiento son responsabilidad del usuario. No enfriar.
Niños
Pacientes de 12 a 17 años de edad
La dosificación es la misma para adultos y niños de 12 a 17 años de edad.
Pacientes menores de 12 años de edad
Tratamiento «según necesidad»
El cálculo de la dosis necesaria del factor IX para pacientes menores de 12 años se basa en la observación empírica de que la administración de una unidad internacional (UI) de factor IX por cada kg de peso corporal aumenta la actividad del factor IX en plasma en 0,7 UI/dl (rango de 0,31 a 1,0 UI/dl) o en un 0,7 % de la actividad normal.
La dosis necesaria se determina mediante la fórmula indicada a continuación.
| Cantidad necesaria de unidades |
= |
masa corporal (kg) |
× |
aumento deseado del nivel del factor IX (% o U/ml) |
× |
inverso del valor de recuperación de actividad registrada (ml/kg) |
Para un aumento progresivo de la recuperación del nivel de actividad de 0,7 UI/ml por 1 UI/kg, la dosis se calcula según la siguiente fórmula:
| Cantidad necesaria de unidades |
= |
masa corporal (kg) |
× |
aumento deseado del nivel de factor IX (% o U/ml) |
× |
1,4 ml/kg |
Para determinar la dosis durante episodios de hemorragia y procedimientos quirúrgicos, se puede utilizar la misma tabla que para adultos (ver tabla 7 anterior).
Prevención
El rango de dosis recomendadas en el tratamiento de pacientes pediátricos menores de 12 años oscila entre 40 y 80 UI/kg cada 3-4 días. En algunos casos, dependiendo de la farmacocinética, la edad, el fenotipo de la hemorragia y el grado de actividad física del paciente, puede ser necesario acortar los intervalos entre administraciones o aumentar las dosis del medicamento.
Sobredosis.
No se han descrito los efectos de dosis más altas que las recomendadas del medicamento RIXUBIS.
Reacciones adversas
Se han observado casos raros de hipersensibilidad o reacciones alérgicas (que pueden incluir angioedema, sensación de ardor y prurito en el lugar de la infusión, escalofríos, enrojecimiento facial, urticaria generalizada, cefalea, erupción cutánea, hipotensión, letargo, náuseas, estado de ansiedad, taquicardia, opresión en el pecho, tinnitus, vómitos, sibilancias), que en ocasiones pueden progresar a una anafilaxia grave (incluyendo shock), ocurrida en estrecha relación temporal con la administración de inhibidores del factor IX.
Después de la estimulación de la inducción de tolerancia inmune en pacientes con hemofilia B y antecedentes de reacciones alérgicas, en quienes estaban presentes inhibidores del factor IX, se han registrado casos de síndrome nefrótico.
Muy raramente se han observado casos de formación de anticuerpos contra proteínas de hámster, asociados con la aparición de reacciones de hipersensibilidad.
En pacientes con hemofilia B pueden desarrollarse anticuerpos neutralizantes (inhibidores) contra el factor IX, lo que se manifiesta por una eficacia terapéutica insuficiente. En tales casos, se recomienda consultar a un centro especializado en el tratamiento de la hemofilia.
El uso de medicamentos basados en el factor IX conlleva un riesgo potencial de episodios tromboembólicos, siendo mayor el riesgo con el uso de productos de bajo grado de purificación. El uso de productos de factor IX de bajo grado de purificación se ha asociado con casos de infarto de miocardio, coagulación intravascular diseminada, trombosis venosa y embolia pulmonar. El uso de productos de factor IX de alto grado de purificación rara vez se asocia con la aparición de tales reacciones adversas.
Lista de reacciones adversas en forma de tabla
En estudios clínicos sobre el uso del medicamento RIXUBIS, participaron 99 personas con al menos una administración del producto, lo que estuvo asociado con un total de 5 reacciones adversas. La tabla que figura a continuación contiene información sobre las reacciones adversas clasificadas por sistemas y órganos (SOC) según la clasificación MedDRA (utilizando términos de uso preferente).
La frecuencia de las reacciones adversas se clasificó según el siguiente criterio: muy frecuentes (> 1/10), frecuentes (de > 1/100 a < 1/10), poco frecuentes (de > 1/1000 a < 1/100), raras (de > 1/10000 a < 1/1000), muy raras (< 1/10000), frecuencia desconocida (con los datos disponibles no es posible estimar la frecuencia).
Dentro de cada categoría de frecuencia, las reacciones adversas se presentan en orden decreciente de gravedad.
Tabla 8
| Reacciones adversas registradas en ensayos clínicos y notificaciones espontáneas |
||
| Clase de sistemas y órganos según la clasificación MedDRA |
Reacciones adversas |
Frecuencia, por paciente |
| Trastornos del sistema inmunitario |
Hipersensibilidad a) |
No conocido |
| Trastornos del sistema nervioso |
Alteración del gusto |
Frecuente |
| Alteraciones del sistema musculoesquelético y del tejido conjuntivo |
Dolor en extremidades |
Frecuente |
a) PR (reacción adversa) se explica a continuación en la sección.
Descripción de reacciones adversas individuales
Hipersensibilidad
Las reacciones de tipo alérgico se manifestaron como disnea, prurito, urticaria generalizada y erupción cutánea.
Niños
Se espera que la frecuencia, tipo y gravedad de las reacciones adversas en niños sean similares a las de los adultos. Sin embargo, no existen datos disponibles para pacientes no tratados previamente, ya que en los estudios clínicos solo se incluyeron pacientes que habían recibido tratamiento anterior. Debido a esto, no se han realizado estudios sobre inmunogenicidad en relación con la formación de inhibidores en pacientes de este grupo de riesgo.
Notificación de reacciones adversas sospechosas
La notificación de reacciones adversas sospechosas tras la autorización del medicamento es de gran importancia. Permite continuar con el seguimiento de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben informar sobre todos los casos de reacciones adversas sospechosas y falta de eficacia del medicamento a través del Sistema de Información Automatizado de Farmacovigilancia en el siguiente enlace: https://aisf.dec.gov.ua.
Período de validez.
3 años.
Condiciones de conservación.
Conservar a una temperatura no superior a 30 °C. No congelar.
Conservar en un lugar inaccesible para los niños.
Incompatibilidades.
Debido a la falta de estudios sobre compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
Al utilizar este medicamento, solo deben emplearse jeringas de plástico con cono de acoplamiento tipo Luer. Debido a la absorción del factor IX de la coagulación sanguínea humana en las superficies internas de ciertos equipos de infusión, la dosificación del medicamento podría no ser precisa.
Envase.
1 vial con polvo (250 UI, 500 UI, 1000 UI, 1500 UI, 2000 UI o 3000 UI), acompañado de 1 vial con disolvente (5 ml de agua para inyección) y 1 dispositivo de reconstitución BAXJECT II, en una caja.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
Baxalta Belgium Manufacturing SA / Baxalta Belgium Manufacturing SA.
Dirección del fabricante y lugar de ejercicio de su actividad.
Boulevard Rene Branquart 80, Lessines, 7860, Bélgica / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.