Ofev®
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO OFEV® (OFEV®)
Composición:
Principio activo: nintedanib;
1 cápsula contiene 100 mg o 150 mg de nintedanib (en forma de esilato);
Sustancias auxiliares: triglicéridos de cadena media, grasa sólida, lecitina (de soja) (E 322);
Vaina de la cápsula: gelatina, glicerol al 85%, dióxido de titanio (E 171), óxido de hierro rojo (E 172), óxido de hierro amarillo (E 172);
Tinta negra para la impresión de las cápsulas: laca, etanol, propilenglicol (E 1520), óxido de hierro negro (E 172).
Forma farmacéutica. Cápsulas blandas.
Principales propiedades físico-químicas:
OFEV, cápsulas blandas de 100 mg
Cápsulas gelatinosas blandas, alargadas, opacas, de color melocotón, con el logotipo de la empresa «Boehringer Ingelheim» y la impresión «100» en tinta negra en un lado.
Las cápsulas contienen una suspensión viscosa de color amarillo brillante.
OFEV, cápsulas blandas de 150 mg
Cápsulas gelatinosas blandas, alargadas, opacas, de color marrón, con el logotipo de la empresa «Boehringer Ingelheim» y la impresión «150» en tinta negra en un lado.
Las cápsulas contienen una suspensión viscosa de color amarillo brillante.
Grupo farmacoterapéutico. Agentes antineoplásicos. Inhibidores de la proteína quinasa.
Código ATC L01EX09.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
Nintedanib es un inhibidor de tirosina quinasa de bajo peso molecular que bloquea los receptores, incluidos el receptor del factor de crecimiento derivado de plaquetas (PDGFR) α y β, el receptor del factor de crecimiento de fibroblastos (FGFR) 1-3 y el receptor del factor de crecimiento endotelial vascular (VEGFR) 1-3. Además, nintedanib inhibe las quinasas Lck (quinasa de tirosina específica de linfocitos), Lyn (quinasa de proteína tirosina), Src (protooncogénica quinasa de tirosina proteica) y CSF1R (receptor del factor estimulante de colonias 1). Nintedanib interactúa competitivamente con el sitio de unión al trifosfato de adenosina (ATP) de estas quinasas y bloquea la transmisión intracelular en cascada de señales, que se ha demostrado que participa en la patogénesis del remodelado del tejido fibroso en enfermedades pulmonares intersticiales.
Efectos farmacodinámicos
En estudios in vitro utilizando células humanas, se ha demostrado que nintedanib inhibe procesos implicados en la iniciación del proceso fibrogénico, la liberación de mediadores profibróticos desde monocitos de sangre periférica y la polarización de macrófagos hacia macrófagos activados alternativamente. Se ha demostrado que nintedanib suprime procesos fundamentales en la fibrosis orgánica, como la proliferación y migración de fibroblastos, así como la transformación en fenotipo activo de los miofibroblastos y la secreción de matriz extracelular. En estudios en animales en varios modelos de fibrosis pulmonar idiopática (FPI), enfermedad mixta del tejido conectivo (EMTC)/fibrosis pulmonar asociada a EMTC, enfermedad del tejido conectivo (ETC), artritis reumatoide (AR) asociada a ETC y fibrosis de otros órganos, nintedanib demostró actividad antiinflamatoria y antifibrótica en pulmón, piel, corazón, riñón e hígado. Nintedanib también mostró actividad vascular. Redujo el apoptosis endotelial microvascular dérmico y atenuó el remodelado vascular pulmonar mediante la reducción de la proliferación de células musculares lisas vasculares, el grosor de la pared vascular pulmonar y la proporción de vasos pulmonares ocluidos.
Eficacia y seguridad clínicas
Fibrosis pulmonar idiopática (FPI)
La eficacia clínica de nintedanib fue evaluada en pacientes con FPI en dos estudios aleatorizados, doble ciego, controlados con placebo, de fase III con diseño idéntico (INPULSIS-1 (1199,32) e INPULSIS-2 (1199,34)). Se excluyeron del estudio los pacientes con un valor basal calculado de CVF < 50 % o una capacidad de difusión para el monóxido de carbono (DLCO) corregida por hemoglobina < 30 %, calculada en el valor basal. Los pacientes fueron aleatorizados en una proporción 3:2 al grupo del medicamento OFEV 150 mg o al grupo placebo, con administración del fármaco dos veces al día durante 52 semanas.
El punto final primario fue la tasa anual de declive del volumen espiratorio forzado en un segundo (CVF). Los puntos finales secundarios clave fueron el cambio en el puntaje total del cuestionario del Hospital Saint George para la evaluación de la función respiratoria (SGRQ) en la semana 52 respecto al valor basal, así como el tiempo hasta la primera exacerbación de la FPI.
Tasa anual de declive del CVF
La tasa anual de declive del CVF (en ml) mostró una reducción significativa en los pacientes que recibieron nintedanib en comparación con aquellos que recibieron placebo. El efecto terapéutico fue similar en ambos estudios (ver Tabla 1).
Tabla 1
Tasa anual de declive del CVF en los estudios INPULSIS-1, INPULSIS-2 y datos combinados en la población de pacientes tratados
| Estudio |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2, datos combinados |
|||
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
204 |
309 |
219 |
329 |
423 |
638 |
| Cambio (EE) en el volumen espiratorio forzado en 1 segundo (VEF1) a las 52 semanas |
−239,9 |
−114,7 |
−207,3 |
−113,6 |
−223,5 |
−113,6 |
| (18,71) |
(15,33) |
(19,31) |
(15,73) |
(13,45) |
(10,98) |
|
| Comparación con placebo |
||||||
| Diferencia1 |
125,3 |
93,7 |
109,9 |
|||
| IC del 95 % |
(77,7, |
(44,8, |
(75,9, |
|||
| 172,8) |
142,7) |
144,0) |
||||
| valor p |
< 0,0001 |
0,0002 |
< 0,0001 |
|||
| 1 Evaluado según un modelo de regresión con coeficientes aleatorios. IC – intervalo de confianza. EE – error estándar. |
En un análisis de sensibilidad que asumió que en los pacientes con datos faltantes en la semana 52 la reducción del VEF1 desde el último valor registrado fue similar a la observada en todos los pacientes que recibieron placebo, la diferencia ajustada en la tasa anual de declive del VEF1 entre los grupos de nintedanib y placebo fue de 113,9 ml/año (IC del 95 % 69,2; 158,5) en el estudio INPULSIS-1 y de 83,3 ml/año (IC del 95 % 37,6; 129,0) en el estudio INPULSIS-2.
En la Fig. 1 se muestra la evolución temporal respecto al valor basal en ambos grupos de tratamiento, basada en un análisis agregado de los datos obtenidos en los estudios INPULSIS-1 e INPULSIS-2.
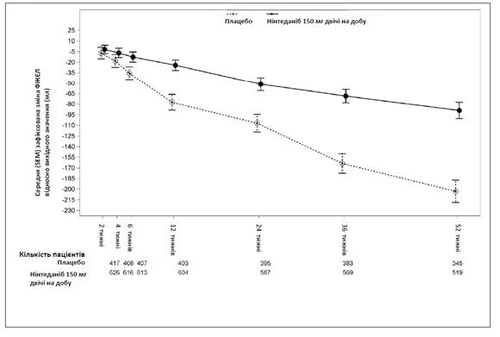
Fig. 1. Cambio medio (EE) del VEF1 registrado a lo largo del tiempo respecto al valor basal (ml), datos combinados de los estudios INPULSIS-1 e INPULSIS-2.
Análisis de los datos de los pacientes respondedores según el parámetro del VEF1
En ambos estudios INPULSIS, el porcentaje de pacientes respondedores según el parámetro del VEF1, categoría en la que se incluyeron aquellos pacientes cuya reducción absoluta calculada del VEF1 en % no superó el 5 % (umbral que indica un mayor riesgo de mortalidad en la FPI), fue significativamente mayor en el grupo de nintedanib que en el grupo de placebo. Resultados similares se observaron en el análisis utilizando el umbral tradicional del 10 % (ver Tabla 2).
Tabla 2
Porcentaje de pacientes respondedores según el parámetro del VEF1 en la semana 52 en los estudios INPULSIS-1, INPULSIS-2 y datos combinados en la población de pacientes tratados
| Estudio |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2, datos combinados |
|||
| Tratamiento |
Placebo |
OFEV, 150 mg dos veces al día |
Placebo |
OFEV, 150 mg dos veces al día |
Placebo |
OFEV, 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
204 |
309 |
219 |
329 |
423 |
638 |
| Valor límite del 5 % |
||||||
| Número (%) de pacientes que respondieron al tratamiento según el parámetro de CVF |
78 (38,2) |
163 (52,8) |
86 (39,3) |
175 (53,2) |
164 (38,8) |
338 (53,0) |
| En comparación con placebo |
||||||
| Relación de probabilidad |
1,85 |
1,79 |
1,84 |
|||
| IC del 95 % |
(1,28; 2,66) |
(1,26; 2,55) |
(1,43; 2,36) |
|||
| Valor de p2 |
0,0010 |
0,0011 |
<0,0001 |
|||
| Valor límite del 10 % |
||||||
| Número (%) de pacientes que respondieron al tratamiento según el parámetro de CVF |
116 (56,9) |
218 (70,6) |
140 (63,9) |
229 (69,6) |
256 (60,5) |
447 (70,1) |
| En comparación con placebo |
||||||
| Relación de probabilidad |
1,91 |
1,29 |
1,58 |
|||
| IC del 95 % |
(1,32; 2,79) |
(0,89; 1,86) |
(1,21; 2,05) |
|||
| Valor de p2 |
0,0007 |
0,1833 |
0,0007 |
|||
1 Pacientes que respondieron al tratamiento: pacientes con una reducción absoluta del VEF1 no superior al 5 % o al 10 % del valor calculado del VEF1 en %, según el umbral aplicable, con evaluación del VEF1 en la semana 52.
2 Basado en regresión logística.
Tiempo hasta la progresión de la enfermedad (reducción absoluta del valor calculado del VEF1 en % ≥ 10 % o muerte)
En ambos estudios INPULSIS se demostró una reducción clínicamente significativa del riesgo de progresión de la enfermedad en pacientes que recibieron tratamiento con nintedanib, en comparación con aquellos que recibieron placebo. En el análisis agrupado, la razón de riesgos fue de 0,60, lo que indica una reducción del 40 % del riesgo de progresión de la enfermedad en los pacientes tratados con nintedanib en comparación con los que recibieron placebo.
Tabla 3
Porcentaje de pacientes con una reducción absoluta del valor calculado del VEF1 en % ≥ 10 % o muerte (eventos) durante 52 semanas y tiempo hasta la progresión de la enfermedad en los estudios INPULSIS-1, INPULSIS-2 y datos agrupados en la población de pacientes tratados
| Estudio |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2, datos combinados |
|||||
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
||
| Número de pacientes en riesgo |
204 |
309 |
219 |
329 |
423 |
638 |
||
| Pacientes con eventos, N (%) |
83 |
75 |
92 |
98 |
175 |
173 |
||
| (40,7) |
(24,3) |
(42,0) |
(29,8) |
(41,4) |
(27,1) |
|||
| Comparación con placebo1 |
||||||||
| Valor p2 |
0,0001 |
0,0054 |
< 0,0001 |
|||||
| Relación de riesgos3 |
0,53 |
0,67 |
0,60 |
|||||
| IC del 95 % |
(0,39; 0,72) |
(0,51; 0,89) |
(0,49; 0,74) |
|||||
| 1 Basado en datos recopilados durante el período hasta 372 días (52 semanas + 7 días). 2 Basado en la prueba de rango logarítmico. 3 Basado en el modelo de regresión de Cox. |
||||||||
Cambio en el puntaje total del SGRQ en la semana 52 respecto al valor inicial
En el análisis agrupado de los estudios INPULSIS, los valores basales del SGRQ fueron de 39,51 en el grupo de nintedanib y de 39,58 en el grupo de placebo. El cambio promedio estimado en el puntaje total del SGRQ en la semana 52 respecto al valor inicial fue menor en el grupo de nintedanib (3,53) que en el grupo de placebo (4,96), con una diferencia entre los grupos de tratamiento de -1,43 (IC del 95 %: -3,09; 0,23; p = 0,0923). En general, el efecto de nintedanib sobre la calidad de vida relacionada con la salud, medida mediante el puntaje total del SGRQ, fue modesto y mostró una menor progresión del deterioro en comparación con el placebo.
Tiempo hasta la primera exacerbación de la EFP
En el análisis agrupado de los estudios INPULSIS, los pacientes que recibieron nintedanib presentaron un riesgo numéricamente menor de primera exacerbación en comparación con aquellos que recibieron placebo (ver tabla 4).
Tabla 4
Porcentaje de pacientes con exacerbaciones de la EFP (eventos) durante el periodo de 52 semanas y tiempo hasta la primera exacerbación basado en datos informados por el investigador, en los estudios INPULSIS-1, INPULSIS-2 y datos agrupados en la población de tratamiento
| Estudio |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2, datos combinados |
|||||
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
||
| Cantidad de pacientes en riesgo |
204 |
309 |
219 |
329 |
423 |
638 |
||
| Pacientes con eventos, N (%) |
11 (5,4) |
19 (6,1) |
21 (9,6) |
12 (3,6) |
32 (7,6) |
31 (4,9) |
||
| Comparación con placebo1 |
||||||||
| Valor p2 |
0,6728 |
0,0050 |
0,0823 |
|||||
| Relación de riesgos3 riesgos3 |
1,15 |
0,38 |
0,64 |
|||||
| IC del 95 % |
(0,54; 2,42) |
(0,19; 0,77) |
(0,39; 1,05) |
|||||
| 1 Basado en datos recopilados durante el período hasta los 372 días (52 semanas + 7 días). 2 Basado en la prueba de rangos logarítmicos. 3 Basado en el modelo de regresión de Cox. |
||||||||
El análisis de sensibilidad mostró que el porcentaje de pacientes con al menos un episodio de exacerbación evaluado por expertos durante las 52 semanas fue menor en el grupo de nintedanib (1,9 % de los pacientes) que en el grupo placebo (5,7 % de los pacientes). En el análisis del tiempo hasta la aparición del primer episodio de exacerbación evaluado, utilizando datos agrupados, se obtuvo un índice de riesgo (HR) de 0,32 (IC del 95 %: 0,16, 0,65; p = 0,0010). Esto indica que el riesgo de presentar la primera exacerbación de la FPI fue estadísticamente significativo menor en el grupo de nintedanib que en el grupo placebo, en cualquier momento del tiempo.
Análisis de supervivencia
En el análisis agrupado de los datos de supervivencia según variables predefinidas en los estudios INPULSIS, la mortalidad global durante el periodo de 52 semanas fue menor en el grupo de nintedanib (5,5 %) que en el grupo placebo (7,8 %). En el análisis del tiempo hasta la muerte se determinó un HR de 0,70 (IC del 95 %: 0,43, 1,12; p = 0,1399). Los resultados de todos los parámetros de punto final relacionados con la supervivencia (como mortalidad durante el tratamiento y mortalidad por eventos respiratorios) mostraron diferencias numéricas consistentes a favor de nintedanib.
Tabla 5
Mortalidad por todas las causas (eventos) durante el periodo de 52 semanas en los estudios INPULSIS-1,
INPULSIS-2 y datos agrupados en la población de pacientes tratados
| Estudio |
INPULSIS-1 |
INPULSIS-2 |
INPULSIS-1 e INPULSIS-2, datos combinados |
|||
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
Placebo |
OFEV 150 mg dos veces al día |
| Número de pacientes en riesgo |
204 |
309 |
219 |
329 |
423 |
638 |
| Pacientes con eventos, N (%) |
13 (6,4) |
13 (4,2) |
20 (9,1) |
22 (6,7) |
33 (7,8) |
35 (5,5) |
| Comparación con placebo1 |
||||||
| Valor p2 |
0,2880 |
0,2995 |
0,1399 |
|||
| Relación de riesgos3 |
0,63 |
0,74 |
0,70 |
|||
| IC del 95 % |
(0,29; 1,36) |
(0,40; 1,35) |
(0,43; 1,12) |
|||
| 1 Basado en datos recopilados durante un período de hasta 372 días (52 semanas + 7 días). 2 Basado en la prueba de rango logarítmico. 3 Basado en el modelo de regresión de Cox. |
Tratamiento prolongado con Ofev en pacientes con FPI (INPULSIS-ON)
En un estudio abierto de extensión con Ofev participaron 734 pacientes con FPI. Los pacientes que completaron el período de tratamiento de 52 semanas en el estudio INPULSIS recibieron tratamiento abierto con Ofev en el estudio de extensión INPULSIS-ON. La mediana de duración del tratamiento para los pacientes que recibieron Ofev en ambos estudios (INPULSIS y INPULSIS-ON) fue de 44,7 meses (rango: 11,9–68,3). Los parámetros de eficacia evaluados incluyeron las tasas anuales de declive de la CVF durante 192 semanas, que fue de -135,1 (5,8) ml/año en todos los pacientes tratados, lo que corresponde a la tasa anual de disminución de la CVF observada en los pacientes que recibieron Ofev en el estudio de fase III INPULSIS (-113,6 ml/año). El perfil de seguridad de Ofev en el estudio INPULSIS-ON fue coherente con el perfil de seguridad observado en el estudio de fase III INPULSIS.
Pacientes con FPI y alteración grave de la función pulmonar (INSTAGE)
INSTAGE fue un estudio clínico prospectivo, aleatorizado, multicéntrico, multinational, doble ciego, con grupos paralelos y una duración de 24 semanas, que incluyó pacientes con FPI y alteración grave de la función pulmonar (DLCO ≤ 35 %). Un total de 136 pacientes recibieron Ofev como monoterapia. El resultado correspondiente al criterio principal mostró una reducción del puntaje total del cuestionario del Hospital Saint George para la evaluación de la función respiratoria (SGRQ) de -0,77 unidades a la semana 12, basado en el cambio medio ajustado respecto al valor basal. Un análisis comparativo a posteriori mostró que la disminución del volumen espiratorio forzado (VEF) en estos pacientes fue comparable a la disminución del VEF observada en pacientes con enfermedad menos avanzada que recibieron Ofev durante el estudio de fase III INPULSIS.
El perfil de seguridad y tolerabilidad de Ofev en pacientes con FPI y alteración grave de la función pulmonar fue coherente con el observado en el estudio de fase III INPULSIS.
Datos adicionales del estudio de fase IV INJOURNEY, en el que se utilizó Ofev a una dosis de 150 mg dos veces al día y pirfenidona como terapia adicional
El tratamiento combinado con nintedanib y pirfenidona fue evaluado en un estudio exploratorio, abierto y aleatorizado de 12 semanas, en el que se administró nintedanib a una dosis de 150 mg dos veces al día junto con pirfenidona como terapia adicional (con titulación de la dosis hasta 801 mg tres veces al día), en comparación con nintedanib a una dosis de 150 mg dos veces al día como monoterapia, en 105 pacientes aleatorizados. El criterio principal fue el porcentaje de pacientes que presentaron eventos adversos gastrointestinales a las 12 semanas respecto al valor basal. Dichos eventos fueron frecuentes, lo cual fue coherente con el perfil de seguridad establecido de cada componente. Las reacciones adversas más frecuentes fueron diarrea, náuseas y vómitos en los pacientes que recibieron pirfenidona junto con nintedanib, en comparación con aquellos que recibieron nintedanib como monoterapia. El cambio medio absoluto de la CVF (capacidad vital forzada) a las 12 semanas respecto al valor basal fue de –13,3 (17,4) ml en los pacientes que recibieron nintedanib y pirfenidona como terapia adicional (n = 48), en comparación con –40,9 (31,4) ml en los pacientes que recibieron nintedanib como monoterapia (n = 44).
Otras enfermedades pulmonares intersticiales fibrosantes crónicas (EPI) con fenotipo progresivo
La eficacia clínica de nintedanib fue evaluada en pacientes con otras EPI fibrosantes crónicas con fenotipo progresivo en un estudio doble ciego, aleatorizado, controlado con placebo, de fase III (INBUILD). Los pacientes con FPI fueron excluidos. Se incluyeron pacientes con diagnóstico clínico de EPI fibrosante crónica si presentaban fibrosis significativa (hallazgos característicos de fibrosis > 10 %) en la TCAR y signos clínicos de progresión (disminución de la CVF ≥ 10 %, disminución de la CVF ≥ 5 % y < 10 % con empeoramiento de síntomas o hallazgos en TCAR, o empeoramiento de síntomas y hallazgos en TCAR durante los 24 meses previos al cribado). Los pacientes debían tener una CVF ≥ 45 % del valor predicho y una DLCO entre 30 % y < 80 % del valor predicho. Los pacientes debían presentar progresión de la enfermedad a pesar del tratamiento, considerado adecuado por los investigadores de acuerdo con la práctica clínica para la EPI correspondiente.
Un total de 663 pacientes fueron aleatorizados en una proporción 1:1 para recibir 150 mg del fármaco dos veces al día o placebo correspondiente durante al menos 52 semanas. La mediana de duración del tratamiento con Ofev durante todo el estudio fue de 17,4 meses, y la duración media del tratamiento fue de 15,6 meses. La aleatorización se estratificó según el patrón de fibrosis en TCAR determinado por expertos centrales. Se aleatorizaron 412 pacientes con un patrón de fibrosis en TCAR consistente con neumonía intersticial usual (NIU) y 251 pacientes con otros patrones de fibrosis en TCAR. Para el análisis en este estudio se definieron dos poblaciones primarias: todos los pacientes (población total) y los pacientes con un patrón de fibrosis en TCAR consistente con NIU. Los pacientes con otros patrones de fibrosis en TCAR representaron una población adicional.
El criterio principal fue la tasa anual de disminución de la capacidad vital forzada (CVF) (ml) durante 52 semanas. Los criterios secundarios clave fueron el cambio absoluto respecto al valor basal en el cuestionario breve King's Breathlessness in Interstitial Lung Disease (K-BILD) durante 52 semanas, el tiempo hasta la primera exacerbación aguda de EPI o muerte durante 52 semanas y el tiempo hasta la muerte durante 52 semanas.
Los pacientes tenían una edad media (desviación estándar [DE; mín.–máx.]) de 65,8 (9,8; 27–87) años y un valor medio de CVF del 69,0 % del valor predicho (15,6; 42–137). Los principales diagnósticos clínicos de EPI en los grupos incluidos en el estudio fueron neumonitis por hipersensibilidad (26,1 %), EPI autoinmunes (25,6 %), neumonía intersticial inespecífica idiopática (18,9 %), neumonía intersticial idiopática no clasificada (17,2 %) y otras EPI (12,2 %).
Tasa anual de disminución de la CVF
La tasa anual de disminución de la CVF (ml) durante 52 semanas mostró una reducción significativa en los pacientes que recibieron Ofev en comparación con aquellos que recibieron placebo (tabla 6), lo que corresponde a una eficacia relativa del tratamiento del 57,0 %.
Tabla 6
Tasa anual de disminución de la CVF (ml) durante 52 semanas
| Parámetros |
Placebo |
OFEV 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
331 |
332 |
| Índice 1 (EE) de disminución durante 52 semanas |
-187,8 (14,8) |
-80,8 (15,1) |
| Comparación con placebo |
||
| Diferencia 1 |
107,0 |
|
| IC del 95 % |
(65,4; 148,5) |
|
| valor de p |
< 0,0001 |
|
1 Sobre la base de una regresión mixta con efectos fijos para el tratamiento, patrones de TCAR, efecto constante fijo del tiempo, valor inicial de la CVF (ml), así como la interacción del tratamiento con el tiempo y del valor inicial con el tiempo.
Resultados similares se observaron en la población secundaria de pacientes con patrón UIP según TCAR. El efecto del tratamiento fue consistente en la población adicional de pacientes con otros patrones de fibrosis según TCAR (valor-\textit{p} de interacción 0,2268) (Fig. 2).
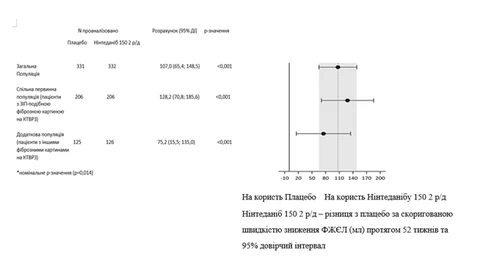
Fig. 2. Diagrama de la tasa anual de declive de la CVF (ml) durante 52 semanas en las poblaciones de pacientes.
El efecto del medicamento OFEV sobre la reducción de la tasa anual de declive de la CVF fue confirmado por todos los análisis de sensibilidad previamente definidos; resultados consistentes se observaron en los subgrupos de eficacia previamente definidos: por sexo, edad, raza, valor inicial de CVF (% del valor predicho) y diagnóstico clínico principal de EIP en los grupos.
En la Fig. 3 se muestra la evolución del cambio en la CVF desde el valor basal en los grupos de tratamiento.
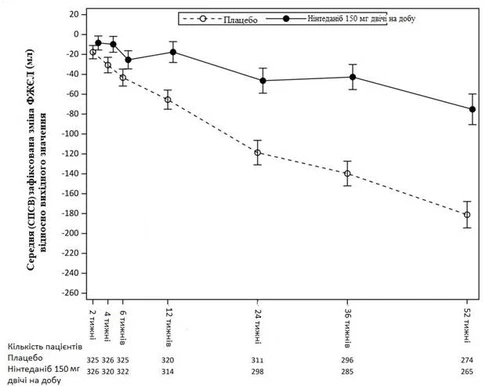
Fig. 3. Cambio medio en la CVF desde el valor basal (ml) ajustado según el modelo de efectos mixtos (SEPM) durante 52 semanas.
Además, se observaron efectos favorables del medicamento OFEV sobre el cambio absoluto medio ajustado en la CVF (% del valor predicho) en comparación con el valor basal a las 52 semanas. El valor medio ajustado del cambio absoluto en la CVF (% del valor predicho) desde el valor basal hasta la semana 52 fue menor en el grupo de nintedanib (-2,62 %) que en el grupo placebo (-5,86 %). La diferencia media ajustada entre los grupos de tratamiento fue de 3,24 (IC 95 %: 2,09; 4,40; \textit{p} nominal < 0,0001).
Análisis de pacientes que alcanzaron el efecto terapéutico según el parámetro de CVF
La proporción de pacientes que alcanzaron el efecto terapéutico según el parámetro de CVF, definido como pacientes con una disminución relativa de la CVF del % del valor predicho no superior al 5 %, fue mayor en el grupo del medicamento OFEV en comparación con el placebo. Resultados análogos se observaron en los análisis que utilizaron un umbral del 10 % (Tabl. 7).
Tabla 7
Tasa anual de declive de la CVF (ml) durante 52 semanas
| Tratamiento |
Placebo |
OFEV, 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
331 |
332 |
| Valor límite del 5 % |
||
| Número (%) de pacientes que alcanzaron el efecto terapéutico según el parámetro VEF1 |
104 (31,4) |
158 (47,6) |
| En comparación con placebo |
||
| Relación de odds2 |
2,01 |
|
| IC del 95 % |
(1,46; 2,76) |
|
| valor p |
< 0,0001 |
|
| Valor límite del 10 % |
||
| Número (%) de pacientes que alcanzaron el efecto terapéutico según el parámetro VEF1 |
169 (51,1) |
197 (59,3) |
| En comparación con placebo |
||
| Relación de odds2 |
1,42 |
|
| IC del 95 % |
(1,04; 1,94) |
|
| valor p |
0,0268 |
|
1 Los pacientes que alcanzaron el efecto terapéutico, evaluado según el parámetro de VEF1, fueron aquellos que no presentaron una disminución relativa del VEF1 (% del valor predicho) mayor del 5 % o mayor del 10 %, dependiendo de los umbrales establecidos, y con evaluación del VEF1 en la semana 52 (los pacientes con datos faltantes en la semana 52 se consideraron como no alcanzaron el efecto terapéutico).
2 Basado en un modelo de regresión logística con la covariable continua del nivel basal de VEF1 (%) y la covariable binaria del patrón de TCAR.
Tiempo hasta la primera exacerbación de EPOC o muerte
Durante todo el estudio, la proporción de pacientes que tuvieron al menos un primer episodio de exacerbación aguda de EPOC o muerte fue del 13,9 % en el grupo OFEV y del 19,6 % en el grupo placebo. La HR fue de 0,67 (IC del 95 %: 0,46; 0,98; p nominal = 0,0387), lo que indica una reducción del 33 % en el riesgo de primera exacerbación de EPOC o muerte en los pacientes que recibieron OFEV en comparación con aquellos que recibieron placebo (fig. 4).
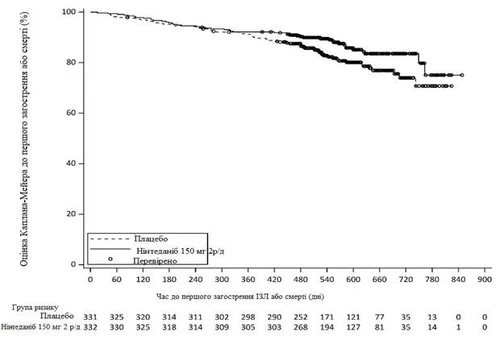
2 v/d – dos veces al día
Fig. 4. Tiempo hasta la primera exacerbación de EPOC o muerte.
Análisis de supervivencia
El riesgo de muerte fue menor en el grupo del fármaco OFEV en comparación con el grupo placebo. La HR fue de 0,78 (IC del 95 %: 0,50; 1,21; p nominal = 0,2594), lo que indica una reducción del 22 % en el riesgo de muerte en los pacientes que recibieron OFEV en comparación con aquellos que recibieron placebo.
Tiempo hasta la progresión (disminución absoluta del VEF1 (% del predicho) ≥ 10 %) o muerte
En el estudio INBUILD, el riesgo de progresión (disminución absoluta del VEF1 (% del predicho) ≥ 10 %) o muerte fue menor en los pacientes que recibieron OFEV. La proporción de pacientes con el evento definido fue del 40,4 % en el grupo del fármaco OFEV y del 54,7 % en el grupo placebo. La HR fue de 0,66 (IC del 95 %: 0,53; 0,83; p = 0,0003), lo que indica una reducción del 34 % en el riesgo de progresión (disminución absoluta del VEF1 (% del predicho) ≥ 10 %) o muerte en los pacientes que recibieron OFEV en comparación con aquellos que recibieron placebo.
Calidad de vida
El cambio medio ajustado desde el valor basal en la puntuación total del K-BILD en la semana 52 fue de -0,79 unidades en el grupo placebo y de 0,55 en el grupo del fármaco OFEV. La diferencia entre los grupos de tratamiento fue de 1,34 (IC del 95 %: -0,31; 2,98; p nominal = 0,1115).
El cambio absoluto medio ajustado desde el valor basal en los síntomas de disnea según el cuestionario L-PF en la semana 52 fue de 4,28 en el grupo del fármaco OFEV frente a 7,81 en el grupo placebo. La diferencia media ajustada entre grupos a favor del fármaco OFEV fue de -3,53 (IC del 95 %: -6,14; -0,92; p nominal = 0,0081). El cambio absoluto medio ajustado desde el valor basal en los síntomas de tos según el cuestionario L-PF en la semana 52 fue de -1,84 en el grupo del fármaco OFEV frente a 4,25 en el grupo placebo. La diferencia media ajustada entre grupos a favor del fármaco OFEV fue de -6,09 (IC del 95 %: -9,65; -2,53; p nominal = 0,0008).
Enfermedad pulmonar intersticial asociada a esclerodermia sistémica (esclerosis sistémica) (EPI-ES)
La eficacia clínica del nintedanib se estudió en pacientes con EPI-ES en un ensayo doble ciego, aleatorizado y controlado con placebo de fase III (SENSCIS). A los pacientes se les diagnosticó EPI-ES según los criterios de clasificación de la ES de la American College of Rheumatology/European League Against Rheumatism de 2013 y mediante tomografía computarizada de alta resolución (TCAR) del tórax en los 12 meses previos. Un total de 580 pacientes fueron aleatorizados en una relación 1:1 para recibir nintedanib (OFÉV) 150 mg dos veces al día o placebo durante un mínimo de 52 semanas. De ellos, 576 pacientes recibieron tratamiento. La aleatorización se estratificó según el estado de los anticuerpos antitopoisomerasa (ATA). Algunos pacientes continuaron el tratamiento en "ciego" hasta las 100 semanas (mediana de exposición a OFÉV: 15,4 meses; exposición media a OFÉV: 14,5 meses).
El punto final primario fue la tasa anual de disminución de la capacidad vital forzada (CVF) durante 52 semanas. Los puntos finales secundarios clave fueron el cambio absoluto desde el valor basal en la piel según la escala de Rodnan modificada (mRSS) en la semana 52 y el cambio absoluto desde el valor basal en la puntuación total del cuestionario del Hospital Saint George para la evaluación de la función respiratoria (SGRQ) en la semana 52.
En la población total, el 75,2 % de los pacientes eran mujeres. La edad media (desviación estándar (DE), mín.-máx.) fue de 54 (12,2; 20–79) años. En total, el 51,9 % de los pacientes tenían la forma sistémica cutánea difusa de esclerosis sistémica (ES); el 48,1 % tenían la forma cutánea limitada de ES. El tiempo medio (DE) desde la aparición del primer síntoma "no Raynaud" fue de 3,49 (1,7) años. El 49,0 % de los pacientes recibían tratamiento estable con micofenolato en el valor basal. El perfil de seguridad en pacientes que recibieron o no micofenolato en el valor basal fue similar.
Tasa anual de disminución del VEF1
La tasa anual de disminución del VEF1 (ml) durante 52 semanas se redujo significativamente en (41,0) ml en los pacientes que recibieron OFÉV en comparación con aquellos que recibieron placebo (tabla 8), lo que corresponde a una eficacia relativa del tratamiento del 43,8 %.
Tabla 8
Tasa anual de disminución del VEF1 (ml) durante 52 semanas
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
288 |
287 |
| Índice 1 (EE) de disminución durante 52 semanas |
-93,3 (13,5) |
-52,4 (13,8) |
| Comparación con placebo |
||
| Diferencia 1 |
41,0 |
|
| IC del 95 % |
(2,9; 79,0) |
|
| valor p |
< 0,05 |
1 Basado en un modelo de regresión aleatoria de coeficientes con un efecto fijo categórico del tratamiento, estado de ATA, sexo del paciente, un efecto fijo constante del tiempo, el valor basal de la FVC (ml), edad y altura del paciente, así como considerando la dependencia del efecto del tratamiento respecto al tiempo y la dependencia de los cambios respecto al valor basal en el tiempo. Se incluyó un efecto aleatorio para el paciente específico y el tiempo. Los errores internos se modelaron mediante una matriz de covarianza no estructurada. La variabilidad interindividual se modeló mediante una matriz de covarianza con componentes de varianza.
El efecto del fármaco OFEV sobre la reducción de la tasa anual de disminución de la FVC fue similar en los análisis de sensibilidad previamente definidos; no se detectó heterogeneidad en los subgrupos previamente definidos de pacientes (por ejemplo, por edad, sexo y uso de micofenolato).
Además, se observaron efectos análogos respecto a otros puntos finales de la función pulmonar, particularmente el cambio en la FVC en la semana 52 (fig. 5 y tabla 9) desde el valor basal y la tasa prevista de disminución de la FVC en % durante 52 semanas (tabla 10), lo que proporciona una justificación adicional del efecto de OFEV en términos de ralentización de la progresión de la ENL-SS. Asimismo, un menor número de pacientes en el grupo de OFEV presentó una reducción absoluta de la FVC > 5 % (20,6 % en el grupo de OFEV frente a 28,5 % en el grupo placebo, OR = 0,65, p = 0,0287). La reducción relativa de la FVC en ml > 10 % fue similar entre los grupos (16,7 % en el grupo de OFEV frente a 18,1 % en el grupo placebo, OR = 0,91, p = 0,6842). En estos análisis, los valores faltantes de FVC en la semana 52 se imputaron utilizando el peor valor registrado en el paciente durante el tratamiento.
Un análisis exploratorio de los datos hasta las 100 semanas (duración máxima del tratamiento en el estudio SENSCIS) muestra que el efecto del tratamiento con OFEV sobre la ralentización de la progresión de la ENL-SS se mantiene más allá de las 52 semanas.
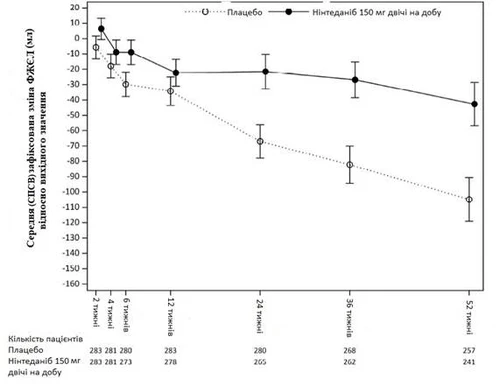
Fig. 5. Cambio medio (IC 95 %) observado en la FVC desde el valor basal (ml) durante 52 semanas.
Tabla 9
Cambio absoluto en la FVC (ml) desde el valor basal en la semana 52
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
288 |
288 |
| Media (DE) en el nivel basal |
2541,0 (815,5) |
2458,5 (735,9) |
| Cambio medio1 (EE) desde el nivel basal a las 52 semanas |
-101,0 (13,6) |
-54,6 (13,9) |
| Comparación con placebo |
||
| Media 1 |
|
|
| IC del 95 % |
(8,1; 84,7) |
|
| valor p |
< 0,05 |
1 Basado en un modelo mixto de medidas repetidas (MMRM) con efectos fijos categóricos del estado de ATA, visita, interacción tratamiento por visita, interacción nivel basal por visita, edad, sexo y altura del paciente. La visita fue una medida repetida. Los errores internos se modelaron mediante una matriz de varianza-covarianza no estructurada. La media ajustada se basó en el número total de pacientes analizados en el modelo (no solo aquellos en los que se realizaron mediciones en el nivel basal y en la semana 52).
Tabla 10
Tasa anual de disminución del VEF1 (%) durante 52 semanas
| Tratamiento |
Placebo |
OFEV 150 mg dos veces al día |
| Número de pacientes cuyos datos fueron analizados |
288 |
287 |
| Índice 1 (EE) de disminución durante 52 semanas |
-2,6 (0,4) |
-1,4 (0,4) |
| Comparación con placebo |
||
| Diferencia 1 |
1,15 |
|
| IC del 95 % |
(0,09; 2,21) |
|
| valor p |
< 0,05 |
1 Basado en una regresión aleatoria de coeficientes con un efecto categórico fijo del tratamiento, estado de ATA, un efecto constante fijo del tiempo, el valor basal de la CVF (% del valor predicho), así como considerando la dependencia del efecto del tratamiento respecto al tiempo y la dependencia de los cambios respecto al nivel basal respecto al tiempo. Se incluyó un efecto aleatorio para el momento específico de registro del paciente y el tiempo. Los errores internos se modelaron mediante una matriz de varianza-covarianza sin estructura. La variabilidad interindividual se modeló mediante una matriz de varianza-covarianza con componentes de varianza.
Cambio desde el valor basal en la puntuación modificada de Rodnan (mRSS) a las 52 semanas
El cambio medio ajustado absoluto desde el valor basal en la mRSS a las 52 semanas fue similar en el grupo del fármaco Ofev (-2,17 (IC del 95 % -2,69, -1,65)) y en el grupo placebo (-1,96 (IC del 95 % -2,48, -1,45)). La diferencia media ajustada entre los grupos de tratamiento fue de -0,21 (IC del 95 % -0,94, 0,53; p = 0,5785).
Cambio desde el valor basal en la puntuación total del cuestionario del Hospital Saint George (SGRQ) a las 52 semanas
El cambio medio ajustado absoluto desde el valor basal en la puntuación total del SGRQ a las 52 semanas fue similar en el grupo del fármaco Ofev (0,81 (IC del 95 % -0,92, 2,55)) y en el grupo placebo (-0,88 (IC del 95 % -2,58, 0,82)). La diferencia media ajustada entre los grupos de tratamiento fue de 1,69 (IC del 95 % -0,73, 4,12; p = 0,1711).
Análisis de supervivencia
La tasa de mortalidad durante todo el período del estudio fue similar en el grupo del fármaco Ofev (N = 10; 3,5 %) y en el grupo placebo (N = 9; 3,1 %). En el análisis del tiempo hasta la muerte durante todo el período del estudio, se determinó un HR de 1,16 (IC del 95 % 0,47, 2,84; p = 0,7535).
Intervalo QT
En el marco de un estudio especial con pacientes con carcinoma de células renales, se realizaron mediciones del intervalo QT/complejo QT; dichas mediciones mostraron que una dosis oral única de 200 mg de nintedanib, así como dosis orales múltiples de 200 mg de nintedanib administradas dos veces al día durante 15 días, no prolongaron el intervalo QT corregido por Fridericia.
Pacientes pediátricos.
Ofev no ha sido estudiado en la práctica pediátrica en la FPI.
Farmacocinética.
Absorción
La concentración máxima de nintedanib en plasma se alcanza aproximadamente entre 2 y 4 horas después de la administración oral del fármaco en forma de cápsulas blandas de gelatina con alimentos (rango de 0,5 a 8 horas). La biodisponibilidad absoluta de la dosis de 100 mg en voluntarios sanos es del 4,69 % (IC del 90 % 3,615–6,078). La absorción y biodisponibilidad se reducen debido a la acción del transportador y al metabolismo presistémico significativo. Se ha demostrado que la exposición a nintedanib aumenta proporcionalmente con la dosis (en rangos de dosis de 50–450 mg una vez al día y 150–300 mg dos veces al día). Se alcanzan concentraciones estables en plasma en un máximo de una semana tras el inicio del tratamiento.
La exposición a nintedanib aumenta aproximadamente en un 20 % si se toma después de las comidas en comparación con la administración en ayunas (IC del 95,3–152,5 %), y la absorción se ralentiza (la mediana del tiempo hasta alcanzar la concentración máxima en plasma en ayunas (tmax) es de 2,00 horas; tras las comidas, de 3,98 horas).
Distribución
La distribución de nintedanib sigue una cinética bifásica. Tras la infusión intravenosa, durante la fase terminal se observa un gran volumen de distribución (Vss): 1050 l, coeficiente geométrico de variación (gCV) del 45,0 %).
La unión de nintedanib a las proteínas plasmáticas humanas in vitro se considera considerable, con una fracción unida del 97,8 %. La albúmina sérica es la principal proteína implicada en la unión. Nintedanib se distribuye principalmente en el plasma, con una relación sangre/plasma de 0,869.
Biotransformación
La reacción principal implicada en el metabolismo de nintedanib es la escisión hidrolítica mediante esterasas, que conduce a la formación del metabolito ácido libre de nintedanib (BIBF 1202). Posteriormente, BIBF 1202 se glucuroniza mediante las enzimas UDP-glucuronosiltransferasa (UGT), específicamente UGT 1A1, UGT 1A7, UGT 1A8 y UGT 1A10, formando el glucurónido de BIBF 1202.
La biotransformación de nintedanib mediante isoformas de CYP es mínima; la isoforma CYP 3A4 desempeña el papel principal en este proceso. En un estudio ADME en humanos, no se detectó en plasma el metabolito principal formado por isoformas de CYP. Según estudios in vitro, el metabolismo dependiente de CYP representa aproximadamente un 5 %, mientras que la escisión mediada por esterasas representa un 25 %. Nintedanib, BIBF 1202 y el glucurónido de BIBF 1202 no inhibieron ni indujeron isoformas de CYP en estudios preclínicos. Por lo tanto, no se esperan interacciones medicamentosas entre nintedanib y sustratos de CYP, inhibidores de CYP o inductores de CYP.
Eliminación
El aclaramiento plasmático total tras infusión intravenosa es alto (1390 ml/min, gCV 28,8 %). La excreción urinaria de la sustancia activa inalterada en 48 horas tras la administración oral de nintedanib es aproximadamente del 0,05 % de la dosis (gCV 31,5 %), y tras administración intravenosa, aproximadamente del 1,4 % (gCV 24,2 %); el aclaramiento renal es de 20 ml/min (gCV 32,6 %). Tras la administración oral de [14C]-nintedanib, el material radiactivo se excreta principalmente por vía biliar y se detecta en las heces (93,4 % de la dosis, gCV 2,61 %). La fracción de excreción renal en el aclaramiento total es baja (0,649 % de la dosis (gCV 26,3 %)). Se considera que la eliminación es completa (más del 90 %) en 4 días tras la administración. El periodo de semivida terminal de nintedanib oscila entre 10 y 15 horas (gCV aproximadamente del 50 %).
Linealidad/no linealidad
Se puede suponer que la farmacocinética (PK) de nintedanib es lineal respecto al tiempo (es decir, los datos de dosis única pueden extrapolarse a datos de uso múltiple). El valor de Cmax debido a la acumulación tras administración múltiple excede en 1,04 veces el valor de Cmax de dosis única, y el valor de AUCτ en 1,38 veces. Las concentraciones mínimas residuales de nintedanib permanecen estables durante más de un año.
Transporte
Nintedanib es sustrato de la glucoproteína P (P-gp). Véase la sección «Interacción con otros medicamentos y otras formas de interacción» para obtener información sobre la posible interacción de nintedanib con este transportador. Se ha demostrado in vitro que nintedanib no es sustrato ni inhibidor de OATP-1B1, OATP-1B3, OATP-2B1, OCT-2 ni MRP-2. Nintedanib tampoco es sustrato de BCRP. In vitro se ha demostrado que nintedanib tiene una actividad inhibitoria débil frente a OCT-1, BCRP y P-gp, que se considera de escasa relevancia clínica. Se ha llegado a la misma conclusión respecto a nintedanib como sustrato de OCT-1.
Farmacocinética en grupos especiales de pacientes
Las propiedades farmacocinéticas de nintedanib fueron comparables en voluntarios sanos, pacientes con FPI, pacientes con otras enfermedades pulmonares intersticiales con fenotipo progresivo, pacientes con EIL-CTD y pacientes con cáncer. En base a los resultados del análisis farmacocinético poblacional en pacientes con FPI y cáncer de pulmón no microcítico (CPNM) (N = 1 191) y estudios descriptivos, factores como el sexo del paciente (ajustado por peso corporal), alteraciones renales leves a moderadas (calculadas según el aclaramiento de creatinina), consumo de alcohol o genotipo de glucoproteína P no influyeron en la acción de nintedanib. El análisis farmacocinético poblacional reveló un efecto moderado del sexo, peso corporal y ascendencia racial sobre la acción de nintedanib, como se describe a continuación. Debido a la alta variabilidad interindividual en la exposición, estos efectos menores no se consideraron clínicamente relevantes (véase la sección «Instrucciones de uso»).
Edad
La exposición a nintedanib aumenta linealmente con la edad. En pacientes de 45 años, el valor de AUCτ,ss fue un 16 % menor, y en pacientes de 76 años, un 13 % mayor, en comparación con pacientes cuya mediana de edad fue de 62 años. El rango de edad evaluado durante el análisis fue de 29 a 85 años; la edad superior a 75 años se observó en aproximadamente el 5 % de la población de pacientes. Según el modelo de análisis farmacocinético poblacional, en pacientes de 75 años o más se observó un aumento de la exposición a nintedanib de aproximadamente un 20–25 % en comparación con pacientes menores de 65 años.
No se realizaron estudios similares con niños.
Peso corporal
Existe una correlación inversa entre el peso corporal y la exposición a nintedanib. En pacientes con un peso corporal de 50 kg (percentil 5), el valor de AUCτ,ss aumentó un 25 %, y en pacientes con un peso corporal de 100 kg (percentil 95), disminuyó un 19 % en comparación con pacientes cuya mediana de peso corporal fue de 71,5 kg.
Raza
La exposición media a nintedanib es un 33–50 % mayor en chinos, taiwaneses e indios y un 16 % mayor en japoneses, y un 16–22 % menor en coreanos, en comparación con pacientes de raza caucásica (ajustado por peso corporal). Los datos sobre pacientes de raza negra son muy limitados; el rango de estos datos es similar al de pacientes de raza caucásica.
Alteración de la función hepática
En un estudio especializado de Fase I en voluntarios con alteraciones leves de la función hepática (Clase A según la escala de Child-Pugh), la exposición basada en Cmax y AUC fue 2,2 veces mayor en comparación con voluntarios sanos (IC del 90 % 1,3–3,7 para Cmax y IC del 90 % 1,2–3,8 para AUC, respectivamente). En voluntarios con alteraciones hepáticas moderadas (Clase B según la escala de Child-Pugh) en comparación con voluntarios sanos, la exposición fue 7,6 veces mayor según Cmax (IC del 90 % 4,4–13,2) y 8,7 veces mayor según AUC (IC del 90 % 5,7–13,1), respectivamente. No se realizó un estudio con pacientes con alteraciones hepáticas graves (Clase C según la escala de Child-Pugh).
Tratamiento concomitante con pirfenidona
En un estudio farmacocinético especial, se estudió la administración concomitante de nintedanib y pirfenidona en pacientes con FPI (fibrosis pulmonar idiopática). El Grupo 1 recibió una dosis única de 150 mg de nintedanib antes y después del aumento de la dosis de pirfenidona hasta 801 mg tres veces al día en estado de equilibrio (N = 20 pacientes tratados). El Grupo 2 recibió tratamiento en estado de equilibrio con 801 mg de pirfenidona tres veces al día y participó en la determinación de parámetros farmacocinéticos antes y después de 7 días de tratamiento concomitante con nintedanib 150 mg dos veces al día (N = 17 pacientes tratados). En el Grupo 1, los valores del cociente ajustado de las medias geométricas [IC del 90 % (IC)] para Cmax y AUC0-tz de nintedanib fueron del 93 % (57–151 %) y del 96 % (70–131 %), respectivamente (n = 12 para comparación intrasujeto). En el Grupo 2, los valores del cociente ajustado de las medias geométricas (IC del 90 %) fueron del 97 % (86–110 %) y del 95 % (86–106 %) para Cmax,ss y AUCτ,ss de pirfenidona, respectivamente (n = 12 para comparación intersujeto).
Este estudio no reveló evidencia de interacción medicamentosa farmacocinética significativa entre nintedanib y pirfenidona cuando se administran en combinación (véase la sección «Instrucciones de uso»).
Tratamiento concomitante con bosentán
En un estudio especial de farmacocinética, se investigó la administración concomitante del medicamento Ofev con bosentán en voluntarios sanos. Los pacientes recibieron una dosis del medicamento Ofev de 150 mg antes y después de varias dosis de bosentán de 125 mg dos veces al día en condiciones de hospitalización. Los cocientes geométricos medios ajustados (IC del 90 % (IC)) fueron del 103 % (86 – 124 %) y del 99 % (91 – 107 %) para Cmax y AUC0-tz de nintedanib, respectivamente (n=13), lo que indica que la administración concomitante de nintedanib con bosentán no modifica la farmacocinética de nintedanib.
Uso concomitante de anticonceptivos hormonales orales
En un estudio farmacocinético especial, pacientes con EIL-CTD recibieron una dosis única de la combinación de 30 mcg de etinilestradiol y 150 mcg de levonorgestrel antes y después de tomar 150 mg de nintedanib dos veces al día durante al menos 10 días. Los cocientes geométricos medios ajustados (IC del 90 % (IC)) fueron del 117 % (108–127 %; Cmax) y del 101 % (93–111 %; AUC0–tz) para etinilestradiol y del 101 % (90–113 %; Cmax) y del 96 % (91–102 %; AUC0–tz) para levonorgestrel, respectivamente (n = 15), lo que indica que la administración concomitante de nintedanib no tiene un efecto significativo sobre los niveles plasmáticos de etinilestradiol y levonorgestrel.
Coeficiente exposición-respuesta
Los análisis del coeficiente exposición-respuesta en pacientes con FPI y otras EIL fibrosantes crónicas con fenotipo progresivo revelaron una relación débil entre los niveles plasmáticos de nintedanib y el aumento de los niveles de ALT y/o AST. La dosis administrada puede ser un mejor predictor del riesgo de diarrea de cualquier intensidad, aunque no se puede excluir el nivel plasmático de nintedanib como factor determinante del riesgo (véase la sección «Instrucciones de uso»).
Características clínicas.
Indicaciones.
OFEV está indicado para el tratamiento de la fibrosis pulmonar idiopática (FPI) en adultos.
OFEV también está indicado para el tratamiento de otras enfermedades pulmonares intersticiales fibrosas crónicas con fenotipo progresivo en adultos (ver sección «Propiedades farmacológicas. Farmacodinamia»).
OFEV está indicado para el tratamiento de la enfermedad pulmonar intersticial asociada a la esclerosis sistémica (esclerodermia sistémica) (EPI-ES).
Contraindicaciones.
Embarazo (ver sección «Uso durante el embarazo o la lactancia**»**).
Hipersensibilidad al nintedanib, al cacahuete o a la soja, o a cualquiera de los excipientes del medicamento.
Interacción con otros medicamentos y otras formas de interacción.
Glicoproteína P (P-gp)
El nintedanib es sustrato de la glicoproteína P (P-gp) (ver sección «Propiedades farmacológicas. Farmacocinética»). En un estudio especial de interacción medicamentosa se ha demostrado que la administración concomitante con ketoconazol, un inhibidor potente de la P-gp, aumenta la exposición al nintedanib en un factor de 1,61 respecto al AUC y en un factor de 1,83 respecto al Cmax. Un estudio especial de interacción medicamentosa mostró que la administración concomitante de rifampicina (un potente inductor de la P-gp) reduce la exposición al nintedanib en un 50,3 % respecto al AUC y en un 60,3 % respecto al Cmax (en comparación con la administración de nintedanib solo). Los inhibidores potentes de la P-gp (por ejemplo, ketoconazol, eritromicina o ciclosporina) pueden aumentar la exposición al nintedanib cuando se administran conjuntamente con OFEV. Por lo tanto, debe vigilarse cuidadosamente la tolerabilidad del nintedanib en los pacientes. En caso de aparición de reacciones adversas, puede ser necesario interrumpir el tratamiento, reducir la dosis o suspender la terapia con OFEV (ver sección «Posología y forma de administración»).
Los inductores potentes de la P-gp (por ejemplo, rifampicina, carbamazepina, fenitoína y preparados a base de hierba de San Juan) pueden reducir la exposición al nintedanib. Se recomienda considerar una terapia concomitante alternativa con ausencia o mínima actividad inductora sobre el sistema P-gp.
Citocromo P450 (CYP)
Los isoenzimas CYP participan solo mínimamente en la biotransformación del nintedanib. En estudios preclínicos, el nintedanib y sus metabolitos (BIBF 1202, metabolito ácido libre del nintedanib, y su glucurónido BIBF 1202) no inhibieron ni indujeron los isoenzimas CYP (ver sección «Propiedades farmacológicas. Farmacocinética»). Por lo tanto, se considera baja la probabilidad de interacciones medicamentosas con nintedanib basadas en el metabolismo CYP.
Administración concomitante con otros medicamentos
La administración concomitante de nintedanib con anticonceptivos hormonales orales no alteró significativamente la farmacocinética de los anticonceptivos orales (ver sección «Propiedades farmacológicas. Farmacocinética»).
La administración concomitante de nintedanib con bosentán no modifica la farmacocinética del nintedanib (ver sección «Propiedades farmacológicas. Farmacocinética»).
Características de la aplicación.
Alteraciones del tracto gastrointestinal
Diarréa
En estudios clínicos (véase la sección «Propiedades farmacológicas. Farmacodinámica»), la diarrea fue el efecto adverso más frecuente del tracto gastrointestinal (véase la sección «Reacciones adversas»). En la mayoría de los pacientes, estos eventos adversos fueron de intensidad leve a moderada y se presentaron durante los primeros 3 meses de tratamiento.
Durante el período poscomercialización, se han notificado casos graves de diarrea que provocan deshidratación y alteraciones electrolíticas. El tratamiento de la diarrea (hidratación adecuada y medicamentos antidiarreicos, por ejemplo, loperamida) debe iniciarse ante los primeros signos de esta. Si aparece diarrea, puede ser necesario reducir la dosis o interrumpir el tratamiento. El tratamiento con OFEV puede reiniciarse a dosis reducida (100 mg dos veces al día) o a dosis completa (150 mg dos veces al día). Si la diarrea grave persiste a pesar del tratamiento sintomático, el tratamiento con OFEV debe suspenderse.
Náuseas y vómitos
Las náuseas y los vómitos fueron eventos adversos frecuentes del tracto gastrointestinal (véase la sección «Reacciones adversas»). En la mayoría de los pacientes, las náuseas y los vómitos fueron de intensidad leve o moderada. En el estudio, las náuseas y los vómitos provocaron la interrupción del tratamiento con OFEV en un 2,1 % y un 1,4 % de los pacientes, respectivamente.
Si los síntomas persisten a pesar del tratamiento sintomático adecuado (incluido el uso de medicamentos antieméticos), puede ser necesario reducir la dosis del medicamento o suspender el tratamiento. El tratamiento puede reiniciarse a dosis reducida (100 mg dos veces al día) o a dosis completa (150 mg dos veces al día). Si los síntomas graves persisten, el tratamiento con OFEV debe suspenderse.
Alteraciones de la función hepática
No se han estudiado la seguridad y eficacia del medicamento OFEV en pacientes con alteraciones de la función hepática moderada (clase B según la escala de Child-Pugh) o grave (clase C según la escala de Child-Pugh). Por lo tanto, no se recomienda el tratamiento de estos pacientes con OFEV (véase la sección «Instrucciones de uso y dosis»). Debido al aumento del efecto del medicamento, el riesgo de reacciones adversas puede incrementarse en pacientes con alteraciones de la función hepática leve (clase A según la escala de Child-Pugh). Para los pacientes con alteraciones de la función hepática leve (clase A según la escala de Child-Pugh), debe administrarse una dosis reducida de OFEV (véanse las secciones «Instrucciones de uso y dosis» y «Propiedades farmacológicas. Farmacocinética»).
Durante el tratamiento con nintedanib se han observado casos de daño hepático inducido por el medicamento, incluyendo daño hepático grave con desenlace letal. La mayoría de los eventos hepáticos ocurren durante los primeros tres meses de tratamiento. Por lo tanto, los niveles de transaminasas hepáticas y bilirrubina deben medirse antes de iniciar el tratamiento y durante el primer mes de tratamiento con OFEV. Posteriormente, estos parámetros deben evaluarse regularmente durante los siguientes dos meses de tratamiento y periódicamente después, por ejemplo, en cada visita del paciente o según indicaciones clínicas.
En la mayoría de los casos, el aumento de los niveles de enzimas hepáticas [ALT, AST, fosfatasa alcalina, gamma-glutamil transferasa (GGT), véase la sección «Reacciones adversas»] y bilirrubina fue reversible tras la reducción de la dosis o la interrupción del tratamiento.
Si los niveles de transaminasas (AST o ALT) aumentan más de 3 veces por encima del límite superior normal, se recomienda reducir la dosis o interrumpir el tratamiento con OFEV y realizar un seguimiento estrecho del paciente. Tan pronto como los niveles de transaminasas regresen al nivel basal, el tratamiento con OFEV puede reiniciarse a dosis completa (150 mg dos veces al día) o a dosis reducida (100 mg dos veces al día), que posteriormente puede aumentarse a la dosis completa (véase la sección «Instrucciones de uso y dosis»). Si el aumento de cualquier parámetro de función hepática se asocia con síntomas clínicos de daño hepático, por ejemplo, ictericia, el tratamiento con OFEV debe suspenderse definitivamente. Deben investigarse causas alternativas del aumento de las enzimas hepáticas.
Los pacientes con bajo peso corporal (menos de 65 kg), de origen asiático y del sexo femenino se encuentran en un grupo de mayor riesgo de aumento de enzimas hepáticas. La exposición al nintedanib aumentó linealmente con la edad del paciente, lo que también incrementa el riesgo de aumento de enzimas hepáticas (véase la sección «Propiedades farmacológicas. Farmacocinética»). Se recomienda un seguimiento cuidadoso de los pacientes con estos factores de riesgo.
Función renal
Durante el tratamiento con nintedanib se han observado casos de alteración de la función renal/insuficiencia renal, algunos de ellos con desenlace letal (véase la sección «Reacciones adversas»).
Durante la terapia con nintedanib se recomienda monitorear el estado de los pacientes, especialmente aquellos con factores de riesgo de alteración de la función renal/insuficiencia renal. En caso de alteración de la función renal/insuficiencia renal, debe considerarse la necesidad de ajustar la dosis (véase la sección «Instrucciones de uso y dosis. Ajuste de la dosis»).
Hemorragias
La inhibición del receptor del factor de crecimiento endotelial vascular (VEGFR) puede estar asociada con un mayor riesgo de hemorragia.
Los estudios clínicos no incluyeron pacientes con riesgo conocido de hemorragia, incluyendo pacientes con predisposición hereditaria a hemorragias o pacientes que recibían terapia anticoagulante en dosis altas. Durante el período poscomercialización se han notificado hemorragias leves y graves, algunas de ellas letales (incluyendo pacientes que recibían terapia anticoagulante u otros medicamentos que podrían provocar hemorragia, así como pacientes sin terapia anticoagulante). Por lo tanto, el tratamiento con OFEV en esta categoría de pacientes solo debe considerarse si el beneficio esperado supera el riesgo potencial.
Tromboembolismo arterial
Los pacientes con infarto de miocardio o accidente cerebrovascular en la historia reciente no participaron en estudios clínicos. En los estudios, los casos de tromboembolismo arterial fueron raros (2,5 % en el grupo OFEV frente a 0,7 % en el grupo placebo en el estudio INPULSIS; 0,9 % en el grupo OFEV frente a 0,9 % en el grupo placebo en el estudio INBUILD; 0,7 % en el grupo OFEV frente a 0,7 % en el grupo placebo en el estudio SENSCIS). En los estudios INPULSIS, la mayoría de los casos de infarto de miocardio ocurrieron en el grupo OFEV (1,6 %) frente al grupo placebo (0,5 %), mientras que las reacciones adversas que reflejan enfermedades isquémicas cardíacas fueron comparables entre los grupos OFEV y placebo. En el estudio INBUILD, el infarto de miocardio fue de baja frecuencia: 0,9 % en el grupo OFEV frente a 0,9 % en el grupo placebo. En el estudio SENSCIS, los casos de tromboembolismo arterial fueron raros: 0,7 % en los pacientes que recibieron placebo y 0,7 % en el grupo que recibió OFEV. El infarto de miocardio fue de baja frecuencia en el grupo placebo (0,7 %) y no se observó en el grupo OFEV. Debe tenerse precaución al tratar pacientes con alto riesgo cardiovascular, incluyendo enfermedad coronaria conocida. Debe considerarse la posibilidad de interrumpir el tratamiento en pacientes que desarrollen síntomas de isquemia miocárdica aguda.
Aneurismas y disección arterial
La administración de inhibidores del factor de crecimiento del endotelio vascular (VEGF) en pacientes con o sin hipertensión puede provocar la formación de aneurismas y/o disección arterial. Antes de iniciar OFEV, debe evaluarse cuidadosamente el riesgo en pacientes con factores de riesgo como hipertensión o antecedentes de aneurisma.
Tromboembolismo venoso
En estudios clínicos no se observó un aumento del riesgo de complicaciones tromboembólicas venosas en pacientes que recibieron nintedanib. Sin embargo, debido al mecanismo de acción del nintedanib, existe un riesgo potencialmente aumentado de eventos tromboembólicos.
Perforaciones del tracto gastrointestinal (GI) y colitis isquémica
En estudios clínicos, la frecuencia de perforaciones fue del 0,3 % en ambos grupos de tratamiento. Sin embargo, debido al mecanismo de acción del nintedanib, puede aumentar el riesgo de perforaciones GI. Durante el período poscomercialización se han notificado casos de perforación GI y colitis isquémica, algunos de ellos letales. Debe tenerse especial cuidado en el tratamiento de pacientes que hayan tenido cirugías abdominales previas, úlcera péptica, enfermedad diverticular o que estén recibiendo corticosteroides o AINEs concomitantemente. Por ello, OFEV solo debe usarse al menos 4 semanas después de cirugías abdominales. Si ocurre una perforación GI o colitis isquémica, el tratamiento con OFEV debe suspenderse. En casos excepcionales, OFEV puede reiniciarse tras una recuperación completa de la colitis isquémica, una evaluación cuidadosa del estado del paciente y otros factores de riesgo.
Proteinuria nefrótica y microangiopatía trombótica
Durante el período poscomercialización se han notificado casos muy raros de proteinuria nefrótica con o sin alteración de la función renal. En algunos casos, los hallazgos histológicos correspondieron a microangiopatía glomerular con o sin trombos renales. Los síntomas desaparecieron tras la suspensión de OFEV, en algunos casos con proteinuria residual. Debe considerarse la conveniencia de suspender el tratamiento en pacientes con signos o síntomas de síndrome nefrótico.
El uso de inhibidores del VEGF se ha asociado con microangiopatía trombótica, incluyendo un número muy reducido de casos con nintedanib. Si los datos clínicos o de laboratorio sugieren microangiopatía trombótica en pacientes que reciben nintedanib, el tratamiento debe suspenderse y realizarse una evaluación cuidadosa.
Hipertensión arterial
El uso de OFEV puede aumentar la presión arterial, por lo que debe medirse periódicamente y según indicaciones clínicas.
Hipertensión pulmonar
Los datos sobre el uso de OFEV en pacientes con hipertensión pulmonar son limitados.
Los pacientes con hipertensión pulmonar significativa (índice cardíaco ≤ 2 l/min/m², epoprostenol/treprostinil parenteral o insuficiencia ventricular derecha significativa) fueron excluidos del estudio INBUILD y SENSCIS. Se recomienda un seguimiento cuidadoso de los pacientes con hipertensión pulmonar.
Alteraciones en la cicatrización de heridas
En estudios clínicos no se observó un aumento en la frecuencia de alteraciones en la cicatrización de heridas. Debido al mecanismo de acción del nintedanib, esta sustancia podría afectar negativamente la cicatrización de heridas. No se han realizado estudios específicos sobre el efecto del nintedanib en la cicatrización de heridas. Por lo tanto, el tratamiento con OFEV debe iniciarse o reiniciarse (si se interrumpió por cirugía) considerando la opinión clínica sobre la adecuada cicatrización de la herida.
Tratamiento de pacientes con EFP progresivas muy raras
El estudio INBUILD no tuvo un diseño ni potencia adecuados para demostrar beneficios del nintedanib en subgrupos con diagnóstico de EFP. Se demostró un efecto consistente en subgrupos basados en diagnósticos de EFP (véase la sección «Farmacodinámica»). La experiencia con el uso de nintedanib en EFP fibrosantes progresivas muy raras es limitada. Los criterios clínicos de progresión utilizados en el estudio INBUILD se describen en la sección «Farmacodinámica».
Terapia concomitante con pirfenidona
En un estudio farmacocinético especial se evaluó el tratamiento concomitante de nintedanib y pirfenidona en pacientes con FPI (fibrosis pulmonar idiopática). Este estudio no mostró evidencia de interacción farmacocinética significativa entre nintedanib y pirfenidona cuando se usan en combinación (véase la sección «Propiedades farmacológicas. Farmacocinética»). Debido a la similitud en los perfiles de seguridad de ambos medicamentos, se podrían esperar reacciones adversas aditivas, incluyendo efectos gastrointestinales y hepáticos. La relación beneficio-riesgo del tratamiento concomitante con pirfenidona no ha sido establecida.
Efecto sobre el intervalo QT
No se han observado signos de prolongación del intervalo QT con el uso de nintedanib en el programa de estudios clínicos (véase la sección «Propiedades farmacológicas. Farmacodinámica»). Dado que se sabe que otros inhibidores de tirosina quinasa afectan el QT, debe administrarse nintedanib con precaución en pacientes en riesgo de prolongación del intervalo QT.
Reacciones alérgicas
Se sabe que los productos nutricionales que contienen soja provocan reacciones alérgicas, incluyendo anafilaxia grave, en personas alérgicas a la soja. Los pacientes con alergia conocida a la proteína de cacahuate se encuentran en un grupo de riesgo de reacciones graves a medicamentos que contienen soja. Una cápsula de 100 mg contiene 1,2 mg de lecitina de soja; una cápsula de 150 mg contiene 1,8 mg de lecitina de soja.
Uso durante el embarazo o la lactancia.
Mujeres en edad fértil
El nintedanib puede tener efectos negativos en el feto humano. Las mujeres en edad fértil deben tomar precauciones para evitar el embarazo y usar métodos anticonceptivos confiables durante el tratamiento con OFEV, al inicio del tratamiento y durante al menos 3 meses después de la última dosis. El nintedanib no afecta significativamente los niveles plasmáticos de etinilestradiol y levonorgestrel (véase la sección «Propiedades farmacológicas. Farmacocinética»). La eficacia de los anticonceptivos hormonales orales puede reducirse en caso de vómitos y/o diarrea u otros estados que puedan afectar la absorción. Se recomienda a las mujeres que toman anticonceptivos hormonales orales y presentan estos estados que usen métodos anticonceptivos alternativos de alta eficacia.
Embarazo
No se han realizado estudios específicos sobre el uso de OFEV en mujeres embarazadas, pero estudios preclínicos en animales han demostrado toxicidad reproductiva de este principio activo. Dado que el nintedanib puede tener efectos embriotóxicos en humanos, no debe usarse durante el embarazo (véase la sección «Contraindicaciones»). Se debe realizar una prueba de embarazo antes de iniciar OFEV y durante el tratamiento según corresponda.
Las pacientes deben informar inmediatamente al médico si se produce un embarazo durante el tratamiento con OFEV.
Si se desarrolla un embarazo durante el tratamiento con OFEV, el tratamiento debe suspenderse y la paciente debe informarse sobre el riesgo potencial de embriotoxicidad del medicamento.
Lactancia
No hay datos sobre la excreción de nintedanib y sus metabolitos en la leche materna humana. En estudios preclínicos, se ha demostrado que pequeñas cantidades de nintedanib y sus metabolitos pasan a la leche materna en animales durante la lactancia (≤ 0,5 % de la dosis administrada). Por lo tanto, no puede descartarse el riesgo para recién nacidos y lactantes. Durante el tratamiento con OFEV, la lactancia debe suspenderse.
Fertilidad
En estudios preclínicos no se observaron signos de alteración de la fertilidad en machos. En estudios de toxicidad subaguda y crónica, en los que los niveles de exposición sistémica fueron comparables a los alcanzados con la dosis máxima recomendada en humanos (150 mg dos veces al día), no se observaron alteraciones de la fertilidad en hembras de animales.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar mecanismos.
OFEV tiene un efecto insignificante sobre la capacidad de conducir vehículos o manejar mecanismos. Durante el uso de OFEV, se debe recomendar a los pacientes tener precaución al conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
El tratamiento con el medicamento debe iniciarse por un médico con experiencia en el manejo de pacientes con enfermedades para las cuales se ha aprobado el uso de OFEV.
Dosis
La dosis recomendada del medicamento es de 150 mg dos veces al día, aproximadamente cada 12 horas. La dosis de 100 mg dos veces al día solo se recomienda para aquellos pacientes que toleran mal la dosis de 150 mg dos veces al día.
Si se omite alguna dosis del medicamento, se debe continuar con la dosis inicialmente recomendada según el horario previsto para la siguiente toma. Si se ha omitido una dosis, el paciente no debe tomar una dosis adicional. La dosis máxima diaria es de 300 mg.
- Ajuste de la dosis *
En caso de aparición de reacciones adversas al medicamento OFEV (ver secciones «Precauciones de uso», «Reacciones adversas»), además del tratamiento sintomático si es necesario, se recomienda reducir la dosis o interrumpir temporalmente el tratamiento hasta que la reacción adversa disminuya a un nivel que permita reanudar la terapia. El tratamiento con OFEV puede reanudarse a la dosis completa (150 mg dos veces al día) o a una dosis reducida (100 mg dos veces al día). Si el paciente no tolera la dosis de 100 mg dos veces al día, el tratamiento con OFEV debe interrumpirse.
Si la diarrea, náuseas y/o vómitos persisten a pesar del tratamiento de apoyo adecuado (incluyendo tratamiento antiemético), puede ser necesario reducir la dosis o interrumpir el tratamiento. El tratamiento con OFEV puede reanudarse a dosis reducida (100 mg dos veces al día) o a la dosis completa (150 mg dos veces al día). En caso de diarrea grave persistente a pesar del tratamiento sintomático, se debe suspender la terapia con OFEV (ver sección «Precauciones de uso»).
En caso de aumento de los niveles de aspartato aminotransferasa (AST) o alanina aminotransferasa (ALT) más de 3 veces por encima del límite superior normal, se recomienda interrumpir el tratamiento con OFEV. Tan pronto como los valores regresen a la normalidad, el tratamiento con OFEV puede reanudarse a dosis reducida (100 mg dos veces al día), que posteriormente puede aumentarse a la dosis completa (150 mg dos veces al día) (ver secciones «Precauciones de uso», «Reacciones adversas»).
Grupos de pacientes especiales
Pacientes de edad avanzada (˃ 65 años)
No se han observado diferencias generales en cuanto a seguridad y eficacia del medicamento en pacientes de edad avanzada. No se requiere ajuste de la dosis según la edad del paciente. A los pacientes de 75 años o más puede ser necesario reducir la dosis para minimizar los efectos adversos (ver sección «Propiedades farmacológicas. Farmacocinética»).
Alteraciones de la función renal
No es necesario ajustar la dosis inicial en pacientes con alteraciones renales leves o moderadas. En pacientes con alteraciones renales graves (aclaramiento de creatinina < 30 ml/min), la seguridad, eficacia y farmacocinética de nintedanib no han sido estudiadas.
Alteraciones de la función hepática
Para pacientes con alteraciones hepáticas leves (Clase A según la escala de Child-Pugh), la dosis recomendada de OFEV es de 100 mg dos veces al día, con intervalos aproximados de 12 horas. En estos pacientes debe considerarse la posibilidad de interrumpir o suspender el tratamiento para controlar las reacciones adversas. En pacientes con alteraciones hepáticas de Clase B y C según la escala de Child-Pugh, la seguridad y eficacia de nintedanib no han sido estudiadas. Por lo tanto, no se recomienda el tratamiento con OFEV en pacientes con alteraciones hepáticas moderadas (Clase B según Child-Pugh) o graves (Clase C según Child-Pugh) (ver sección «Propiedades farmacológicas. Farmacocinética»).
Edad pediátrica
No se han estudiado la seguridad y eficacia del uso de OFEV en niños (menores de 18 años). No hay datos disponibles.
Vía de administración
OFEV está indicado para administración oral. Las cápsulas deben tomarse con alimentos, tragándolas enteras con agua; no deben masticarse. No abrir ni triturar la cápsula. En caso de contacto con el contenido de la cápsula, lávese inmediatamente las manos con abundante agua.
Niños.
El medicamento no se utiliza en la práctica pediátrica.
Sobredosis.
Síntomas
Se han registrado casos de sobredosis en dos pacientes que participaron en un programa oncológico y que tomaron la dosis máxima de 600 mg durante ocho días. Los efectos adversos observados fueron comparables con el perfil de seguridad conocido de nintedanib: aumento de la actividad de las enzimas hepáticas y trastornos gastrointestinales. Ambos pacientes se recuperaron completamente de los efectos adversos. En los estudios INPULSIS se registró un caso de aumento accidental de la dosis por parte de un paciente hasta 600 mg/día durante 21 días. Durante el período de uso incorrecto del medicamento, se observó un solo evento adverso (nasofaringitis) de grado leve, que desapareció durante ese período sin registrar otras reacciones adversas.
Tratamiento
No existe antídoto específico. En caso de sobredosis, debe suspenderse el medicamento y administrarse tratamiento sintomático.
Reacciones adversas.
En los estudios clínicos y durante el período poscomercialización, las reacciones adversas más frecuentes asociadas con el uso de nintedanib fueron diarrea, náuseas y vómitos, dolor abdominal, disminución del apetito, pérdida de peso y aumento de las enzimas hepáticas.
Para obtener información sobre el tratamiento de algunos eventos adversos, véase la sección «Instrucciones de uso».
En la tabla 11 se muestran las reacciones adversas según las clasificaciones por órganos y sistemas de MedDRA y según la frecuencia de aparición, utilizando los siguientes criterios de evaluación:
muy frecuentes (> 1/10); frecuentes (> 1/100 hasta < 1/10); infrecuentes (> 1/1 000 hasta < 1/100); raros (> 1/10 000 hasta < 1/1 000); muy raros (< 1/10 000), desconocido (no puede determinarse con los datos disponibles).
Tabla 11
| Frecuencia |
|||
| Clasificación por órganos y sistemas |
Fibrosis pulmonar idiopática (FPI) |
Enfermedad pulmonar intersticial en esclerodermia sistémica (esclerosis sistémica) (EPIC-ES). |
Otras enfermedades pulmonares intersticiales fibrosantes crónicas (EPIC) con fenotipo progresivo |
| Alteraciones del sistema sanguíneo y linfático |
|||
| Trombocitopenia |
Infrecuente |
Infrecuente |
Infrecuente |
| Alteraciones metabólicas y nutricionales |
|||
| Pérdida de peso |
Frecuente |
Frecuente |
Frecuente |
| Disminución del apetito |
Frecuente |
Frecuente |
Muy frecuente |
| Deshidratación |
Infrecuente |
No conocido |
Infrecuente |
| Alteraciones del corazón |
|||
| Infarto de miocardio |
Infrecuente |
No conocido |
Infrecuente |
| Alteraciones vasculares |
|||
| Hemorragias (ver sección «Precauciones de uso») |
Frecuente |
Frecuente |
Frecuente |
| Hipertensión |
Infrecuente |
Frecuente |
Frecuente |
| Aneurismas y disección arterial |
No conocido |
No conocido |
No conocido |
| Alteraciones del sistema digestivo |
|||
| Diárea |
Muy frecuente |
Muy frecuente |
Muy frecuente |
| Náuseas |
Muy frecuente |
Muy frecuente |
Muy frecuente |
| Dolor abdominal |
Muy frecuente |
Muy frecuente |
Muy frecuente |
| Vómitos |
Frecuente |
Muy frecuente |
Muy frecuente |
| Pancreatitis |
Infrecuente |
No conocido |
Infrecuente |
| Colitis |
Infrecuente |
Infrecuente |
Infrecuente |
| Alteraciones del hígado y de las vías biliares |
|||
| Lesión hepática inducida por el medicamento |
Infrecuente |
Infrecuente |
Frecuente |
| Aumento de las enzimas hepáticas |
Muy frecuente |
Muy frecuente |
Muy frecuente |
| Aumento de alanina aminotransferasa (ALT) |
Frecuente |
Frecuente |
Muy frecuente |
| Aumento de aspartato aminotransferasa (AST) |
Frecuente |
Frecuente |
Frecuente |
| Aumento de la gamma-glutamil transferasa (GGT) |
Frecuente |
Frecuente |
Frecuente |
| Hiperbilirrubinemia |
Infrecuente |
No conocido |
Infrecuente |
| Aumento de fosfatasa alcalina en sangre (FAS) |
Infrecuente |
Frecuente |
Frecuente |
| Alteraciones de la piel y del tejido subcutáneo |
|||
| Erupción cutánea |
Frecuente |
Infrecuente |
Frecuente |
| Picazón |
Infrecuente |
Infrecuente |
Infrecuente |
| Alopecia |
Infrecuente |
No conocido |
Infrecuente |
| Alteraciones renales y de las vías urinarias |
|||
| Insuficiencia renal (ver sección «Precauciones de uso») |
No conocido |
Infrecuente |
No conocido |
| Proteinuria |
Infrecuente |
No conocido |
Infrecuente |
| Alteraciones del sistema nervioso |
|||
| Cefalea |
Frecuente |
Frecuente |
Frecuente |
Descripción de reacciones adversas individuales
Diarrea
En los estudios clínicos (véase la sección «Propiedades farmacológicas. Farmacodinámica»), la diarrea fue el evento adverso más frecuente relacionado con el sistema gastrointestinal. En la mayoría de los pacientes, los eventos adversos fueron de grado leve a moderado. En más de dos tercios de los pacientes, la diarrea se presentó durante los primeros 3 meses de tratamiento.
En la mayoría de los pacientes, los eventos adversos pudieron superarse mediante el uso de terapia antidiarreica, la reducción de la dosis o la interrupción del tratamiento (véase la sección «Instrucciones de uso»). En la tabla 12 se presenta un resumen de los casos de diarrea observados en los estudios clínicos.
Tabla 12
Casos de diarrea en estudios clínicos durante 52 semanas
| Estudio |
INPULSIS |
INBUILD |
SENSCIS |
|||
| Tratamiento |
Placebo |
OFEV |
Placebo |
OFEV |
Placebo |
OFEV |
| Diarréa |
18,4 % |
62,4 % |
23,9 % |
66,9 % |
31,6 % |
75,7 % |
| Diarréa grave |
0,5 % |
3,3 % |
0,9 % |
2,4 % |
1,0 % |
4,2 % |
| Diarréa que provocó la reducción de la dosis de OFEV |
0 % |
10,7 % |
0,9 % |
16,0 % |
1,0 % |
22,2 % |
| Diarréa que provocó la interrupción del tratamiento con OFEV |
0,2 % |
4,4 % |
0,3 % |
5,7 % |
0,3 % |
6,9 % |
Aumento de los niveles de enzimas hepáticas
En los estudios INPULSIS, el aumento de los niveles de enzimas hepáticas (ver sección «Propiedades farmacéuticas») se observó en el 13,6 % de los pacientes tratados con el medicamento OFEV en comparación con el 2,6 % de los pacientes que recibieron placebo. En el estudio INBUILD, el aumento de las enzimas hepáticas se observó en el 22,6 % frente al 5,7 % de los pacientes que recibieron OFEV y placebo, respectivamente. En el estudio SENSCIS, el aumento de los niveles de enzimas hepáticas se observó en el 13,2 % de los pacientes tratados con OFEV en comparación con el 3,1 % de los pacientes que recibieron placebo. El aumento de los niveles de enzimas hepáticas fue reversible y no estuvo asociado con enfermedad hepática clínicamente manifiesta. Información adicional sobre categorías especiales de pacientes, medidas recomendadas y ajuste de dosis en caso de diarrea y aumento de los niveles de enzimas hepáticas se proporciona en las secciones «Propiedades farmacéuticas» y «Posología y forma de administración».
Sangrado
En los estudios clínicos, la proporción de pacientes con eventos de sangrado fue ligeramente mayor o comparable entre los grupos de tratamiento (10,3 % en el grupo de OFEV frente al 7,8 % en el grupo de placebo en el estudio INPULSIS; 11,1 % en el grupo de OFEV frente al 12,7 % en el grupo de placebo en el estudio INBUILD; 11,1 % en el grupo de OFEV frente al 8,3 % en el grupo de placebo en el estudio SENSCIS). Las hemorragias nasales leves fueron los eventos de sangrado más frecuentes notificados como reacciones adversas. Los casos de sangrado grave se notificaron raramente en ambos grupos (1,3 % en el grupo de OFEV frente al 1,4 % en el grupo de placebo en el estudio INPULSIS; 0,9 % en el grupo de OFEV frente al 1,5 % en el grupo de placebo en el estudio INBUILD; 1,4 % en el grupo de OFEV frente al 0,7 % en el grupo de placebo en el estudio SENSCIS).
Durante el período poscomercialización, los casos de sangrado se han relacionado con hemorragias en órganos del sistema digestivo, respiratorio y sistema nervioso central, aunque no se limitan exclusivamente a ellos. La mayor frecuencia de sangrado está asociada al sistema digestivo (ver sección «Propiedades farmacéuticas»).
Proteinuria
En los estudios clínicos, la frecuencia de pacientes con proteinuria fue baja y comparable entre los grupos de tratamiento (0,8 % en el grupo de OFEV frente al 0,5 % en el grupo de placebo en el estudio INPULSIS; 1,5 % en el grupo de OFEV frente al 1,8 % en el grupo de placebo en el estudio INBUILD; 1,0 % en el grupo de OFEV frente al 0,0 % en el grupo de placebo en el estudio SENSCIS). Durante los estudios clínicos no se notificó síndrome nefrótico.
Durante el período poscomercialización se han notificado casos muy raros de proteinuria nefrótica con o sin alteración de la función renal. Los hallazgos histológicos en algunos casos correspondieron a microangiopatía glomerular con o sin trombos renales. La desaparición de los síntomas se observó tras la interrupción del tratamiento con OFEV, en algunos casos con proteinuria residual. Se debe considerar la posibilidad de interrumpir el tratamiento en pacientes que desarrollen signos o síntomas de síndrome nefrótico (ver sección «Propiedades farmacéuticas»).
Notificación de reacciones adversas
La notificación de reacciones adversas tras la autorización del medicamento es importante. Permite una vigilancia continua de la relación beneficio-riesgo del medicamento. Se solicita a los profesionales sanitarios notificar cualquier reacción adversa sospechosa a través del sistema nacional de notificación.
Período de validez. 3 años.
Condiciones de conservación.
Conservar a una temperatura no superior a 25 °C en el envase original para protegerlo de la humedad.
Conservar en un lugar fuera del alcance de los niños.
Envase.
10 cápsulas por blíster de lámina de aluminio con perforación, 6 blísteres por envase de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
Boehringer Ingelheim Pharma GmbH & Co. KG.
Dirección del fabricante y lugar de actividad.
Binger Strasse 173, 55216, Ingelheim am Rhein, Alemania.