Hemax
Ucrania
Contenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO HEMAX (HEMAX)
Composición:
Principio activo: eritropoyetina (epoetina alfa);
1 frasco contiene eritropoyetina (epoetina alfa) 1 000 UI, 2 000 UI, 3 000 UI, 4 000 UI, 10 000 UI, 20 000 UI o 40 000 UI;
Sustancias auxiliares: manitol (E 421), cloruro de sodio, dihidrogenofosfato de sodio dihidratado, hidrogenofosfato de sodio dodecahidratado, albúmina humana.
Forma farmacéutica. Liofilizado para solución inyectable.
Propiedades físico-químicas principales: polvo blanco, homogéneo, compacto.
Grupo farmacoterapéutico. Agentes que actúan sobre el sistema sanguíneo y la hematopoyesis. Fármacos antianémicos. Otros antianémicos. Eritropoyetina.
Código ATC B03X A01.
Propiedades farmacológicas.
Farmacodinamia.
Hemax es un medicamento cuyo ingrediente farmacéutico activo es la epoetina alfa (eritropoyetina humana recombinante, r-HuEPO). La epoetina es una glicoproteína obtenida mediante tecnología recombinante que contiene 165 aminoácidos. Se produce en células de mamífero genéticamente modificadas que contienen el gen de la eritropoyetina. El producto es altamente purificado y contiene una secuencia de aminoácidos idéntica a la eritropoyetina humana natural.
La eritropoyetina induce la eritropoyesis, estimulando la división y diferenciación de las células precursoras eritroides, lo que conduce al aumento del número de eritrocitos y del hematocrito. La eritropoyetina también estimula la liberación de reticulocitos desde la médula ósea hacia la circulación sanguínea, donde maduran hasta convertirse en eritrocitos. Normalmente, la concentración de eritropoyetina en suero oscila entre 10 y 30 UI/ml y está regulada por el nivel de oxigenación de los tejidos. Cuando disminuye el contenido de oxígeno en los tejidos, la concentración de eritropoyetina aumenta entre 100 y 1000 veces. Lo mismo ocurre en las anemias.
Farmacocinética.
Hemax (principio activo: epoetina alfa) se administra por vía parenteral (subcutánea o intravenosa). El contenido de reticulocitos aumenta a las 7–10 días después de la administración del fármaco. El número de eritrocitos, así como los valores de hematocrito y hemoglobina, aumentan generalmente entre las 2 y 6 semanas posteriores a la administración de epoetina alfa. La magnitud y velocidad de la respuesta dependen de la dosis del fármaco y de la disponibilidad de reservas de hierro en suero. La concentración máxima en plasma se alcanza a los 15 minutos tras la administración intravenosa única y entre 5 y 24 horas tras la administración subcutánea única. El nivel máximo de eritropoyetina en suero puede mantenerse entre 12 y 16 horas tras la administración subcutánea, y la eritropoyetina aún se detecta en suero a las 24 horas posteriores a la administración.
El tiempo de semieliminación de la epoetina alfa oscila entre 4 y 13 horas tras la administración intravenosa o subcutánea. El tiempo de semieliminación tras la administración de la primera dosis es más largo que el observado tras dos o más semanas de tratamiento. Normalmente, a las 24 horas el nivel de eritropoyetina en plasma regresa al basal. Tras la administración subcutánea, la concentración máxima en plasma se observa entre 5 y 24 horas, y la posterior disminución de la concentración es más lenta.
En estudios con voluntarios adultos sanos, se ha demostrado que el tiempo de semieliminación tras la administración intravenosa es un 20 % menor que en pacientes con insuficiencia renal. El tiempo de semieliminación tras la administración subcutánea de Hemax en voluntarios adultos sanos es de 20,8 ± 6,3 horas. Tras la interrupción del tratamiento con Hemax, el hematocrito comienza a disminuir a las 2 semanas.
Características clínicas.
Indicaciones.
- Tratamiento de la anemia sintomática asociada a la insuficiencia renal crónica:
- en adultos y niños en hemodiálisis y en adultos en diálisis peritoneal;
- en pacientes adultos con insuficiencia renal que aún no han iniciado hemodiálisis, para el tratamiento de anemia grave de origen renal acompañada de síntomas clínicos.
- Tratamiento de la anemia y reducción del volumen de transfusiones sanguíneas necesarias en pacientes adultos sometidos a quimioterapia por tumores sólidos, linfoma maligno o mieloma múltiple, que presenten un riesgo elevado de transfusión evaluado según el estado general del paciente (incluyendo estado cardiovascular y anemia preexistente antes del inicio de la quimioterapia).
- Hemax está indicado en el marco de un programa de donación previa para facilitar la recolección de sangre autóloga en pacientes con anemia leve (nivel de hemoglobina 10–13 g/dl (6,2–8,1 mmol/l), sin déficit de hierro). El tratamiento solo debe administrarse cuando no sea posible realizar procedimientos ahorradores de sangre o cuando estos sean insuficientes (cuando una intervención quirúrgica planificada de gran envergadura implique una pérdida sanguínea significativa – 4 o más unidades de sangre en mujeres y 5 o más unidades en hombres).
- Hemax está indicado en adultos sin déficit de hierro antes de una intervención quirúrgica ortopédica planificada de gran envergadura con alto riesgo de complicaciones por transfusión, con el fin de reducir el uso de transfusiones sanguíneas alogénicas. El uso del medicamento debe limitarse a adultos con anemia leve (nivel de hemoglobina entre 10–13 g/dl) que no participen en un programa de recolección de sangre autóloga y con una pérdida sanguínea esperada moderada (900–1800 ml).
- Hemax está indicado para el tratamiento de la anemia sintomática (nivel de hemoglobina ≤ 10 g/dl) en pacientes adultos con síndrome mielodisplásico (SMD) de riesgo bajo e intermedio-1, con bajo contenido sérico de eritropoyetina (<200 UI/ml).
Contraindicaciones.
- Hipertensión arterial no controlada;
- desarrollo de aplasia eritroide verdadera (PRCA) como consecuencia del tratamiento con cualquier eritropoyetina (ver sección «Precauciones de uso»);
- hipersensibilidad a la albúmina humana;
- hipersensibilidad a medicamentos obtenidos de células de mamíferos;
- sensibilidad aumentada al principio activo o a cualquiera de los componentes del medicamento;
- enfermedades graves coronarias, periféricas arteriales, carotídeas o cerebrovasculares, así como infarto de miocardio reciente o accidente cerebrovascular en pacientes sometidos a una gran cirugía ortopédica planificada que no hayan participado en un programa de recolección de sangre autóloga;
- imposibilidad de aplicar una profilaxis antitrombótica adecuada en pacientes quirúrgicos;
- contraindicaciones relacionadas con el programa de recolección de sangre autóloga en pacientes que reciben epoetina alfa.
Interacción con otros medicamentos y otras formas de interacción.
No se han observado interacciones del medicamento Hemax con otros fármacos.
No existen datos que indiquen que el tratamiento con epoetina alfa influya en el metabolismo de otros medicamentos.
Los medicamentos que inhiben la eritropoyesis pueden reducir la respuesta al tratamiento con epoetina alfa.
Dado que la ciclosporina se une a los eritrocitos, existe la posibilidad de una interacción medicamentosa. Al administrar conjuntamente Hemax y ciclosporina, se debe controlar el nivel sanguíneo de esta última y, ante un aumento del hematocrito, ajustar la dosis.
No existe evidencia de interacción entre epoetina alfa y el G-CSF (factor estimulante de colonias de granulocitos) o GM-CSF (factor estimulante de colonias de granulocitos y macrófagos) respecto a la diferenciación hematológica o proliferación de células tumorales en muestras de biopsia in vitro.
En mujeres adultas con cáncer de mama metastásico, la administración subcutánea de epoetina alfa a una dosis de 40000 UI/ml simultáneamente con trastuzumab a una dosis de 6 mg/kg no afectó la farmacocinética de trastuzumab.
Características de aplicación.
La presión arterial debe controlarse constantemente en todos los pacientes durante el tratamiento con Hemax. El medicamento debe administrarse con precaución a pacientes con hipertensión no tratada, tratamiento insuficiente de la hipertensión o control inadecuado de la hipertensión. Durante el tratamiento con Hemax puede surgir la necesidad de iniciar o intensificar la terapia antihipertensiva. Si no es posible controlar la presión arterial, debe interrumpirse el uso de epoetina alfa.
Se han observado casos de crisis hipertensivas con encefalopatía y convulsiones que requirieron atención médica inmediata y terapia intensiva, incluso en pacientes con presión arterial normal o baja al inicio del tratamiento. Debe prestarse especial atención a la aparición repentina de cefalea intensa, tipo migraña.
La epoetina alfa debe administrarse con precaución a pacientes con epilepsia, antecedentes de convulsiones o condiciones médicas que representen factores de riesgo para el desarrollo de convulsiones, como infecciones del sistema nervioso central o metástasis cerebral.
La epoetina alfa debe administrarse con precaución a pacientes con insuficiencia hepática crónica. La seguridad del uso de epoetina alfa en esta categoría de pacientes no ha sido establecida.
En pacientes que reciben medicamentos estimulantes de la eritropoyesis, se observa un mayor riesgo de enfermedades vasculares con complicaciones trombóticas, incluyendo trombosis venosa y arterial y embolias (incluyendo casos fatales), tales como trombosis venosa profunda, embolia pulmonar, trombosis de la vena retiniana e infarto de miocardio. También se han notificado casos de accidente cerebrovascular (incluyendo accidente cerebrovascular isquémico, hemorrágico y ataques isquémicos transitorios).
Antes de iniciar el tratamiento con epoetina alfa, deben evaluarse cuidadosamente los riesgos de eventos trombovasculares con complicaciones trombóticas frente al beneficio esperado del uso del medicamento, especialmente en pacientes con factores de riesgo existentes, incluyendo obesidad y antecedentes de eventos trombovasculares (por ejemplo, trombosis venosa profunda, embolia pulmonar e ictus).
Debe controlarse cuidadosamente el nivel de hemoglobina en todos los pacientes debido al riesgo potencialmente aumentado de complicaciones tromboembólicas y muerte si el medicamento se administra con niveles de hemoglobina superiores al rango objetivo indicado.
Durante el tratamiento puede ocurrir un aumento leve y dependiente de la dosis en el número de plaquetas dentro del rango normal. Este parámetro disminuye durante el curso posterior del tratamiento. También se han notificado casos de trombocitosis. Se recomienda controlar regularmente el número de plaquetas durante las primeras 8 semanas de tratamiento.
El nivel de ferritina (o la concentración de hierro en suero) debe determinarse en todos los pacientes antes de iniciar y durante el tratamiento con Hemax. Si es necesario, deben administrarse suplementos de hierro. La falta de respuesta clínica al tratamiento con Hemax requiere investigar posibles causas subyacentes, tales como deficiencia de hierro, ácido fólico o vitamina B12, intoxicación por aluminio, infecciones concurrentes, procesos inflamatorios o traumáticos, hemólisis o fibrosis de médula ósea de cualquier etiología.
Todas las demás causas de anemia (deficiencia de hierro, ácido fólico, vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre, hemólisis o fibrosis de médula ósea de cualquier origen) deben identificarse y tratarse antes de iniciar la terapia con epoetina alfa y antes de decidir aumentar la dosis. En la mayoría de los casos, los valores de ferritina en suero disminuyen simultáneamente con el aumento del hematocrito.
Todos los pacientes tratados con epoetina alfa deben recibir una adecuada suplementación de hierro (200 mg/día por vía oral) durante todo el curso del tratamiento. Para asegurar niveles adecuados de hierro en el organismo, la administración de suplementos de hierro debe iniciarse lo antes posible, incluso varias semanas antes del inicio del programa de recolección de sangre autóloga.
Para garantizar una respuesta óptima al tratamiento con epoetina alfa, es necesario asegurar una ingesta adecuada de hierro:
- A pacientes con insuficiencia renal crónica se recomienda la administración de hierro (200–300 mg/día para adultos y 100–200 mg/día para niños por vía oral, calculado como hierro elemental), si el nivel de ferritina en suero es inferior a 100 ng/ml;
- A pacientes con enfermedades oncológicas se recomienda la administración de hierro (200–300 mg/día por vía oral, calculado como hierro elemental) si la saturación de transferrina es inferior al 20 %;
- A pacientes que participan en un programa de recolección de sangre autóloga se recomienda la administración de hierro (200 mg/día por vía oral, calculado como hierro elemental) varias semanas antes del inicio de la recolección de sangre autóloga con el fin de lograr reservas significativas de hierro antes del inicio del tratamiento y durante el curso del tratamiento con epoetina alfa;
- A pacientes antes de cirugías ortopédicas electivas extensas se recomienda la administración de hierro (200 mg/día por vía oral, calculado como hierro elemental) durante el curso del tratamiento con epoetina alfa. Si es posible, debe iniciarse la administración de hierro antes del inicio del tratamiento con epoetina alfa con el fin de lograr reservas significativas de hierro.
Muy raramente se han notificado casos de desarrollo o empeoramiento de porfiria preexistente en pacientes que recibieron tratamiento con epoetina alfa. La epoetina alfa debe administrarse con precaución a pacientes con porfiria.
Se han notificado casos graves de reacciones adversas relacionadas con el tratamiento con epoetinas, incluyendo el síndrome de Stevens-Johnson y necrólisis epidérmica tóxica, que pueden poner en peligro la vida o tener consecuencias fatales. Los casos más graves se observaron durante el uso de epoetinas de acción prolongada.
Los pacientes deben informarse sobre las posibles reacciones adversas cutáneas. En caso de aparición de síntomas de reacciones cutáneas adversas, debe interrumpirse inmediatamente el tratamiento con epoetina alfa y considerarse métodos alternativos de tratamiento.
Si un paciente desarrolla una reacción cutánea grave durante el tratamiento con epoetina, como el síndrome de Stevens-Johnson o necrólisis epidérmica tóxica, no debe reanudarse en ningún caso el tratamiento con este medicamento.
La denominación comercial de los medicamentos estimulantes de la eritropoyesis utilizados en el tratamiento debe indicarse claramente en la historia clínica del paciente. La transición de un agente estimulante de la eritropoyesis a otro solo es posible bajo supervisión médica.
Anemia eritroide verdadera (PRCA).
Existen informes sobre casos de anemia eritroide verdadera mediada por anticuerpos (PRCA) tras varios meses o años de administración subcutánea de epoetina, principalmente en pacientes con insuficiencia renal crónica. También se han notificado casos de anemia eritroide verdadera en pacientes con hepatitis C que recibieron interferón y ribavirina simultáneamente con agentes estimulantes de la eritropoyesis. La epoetina alfa no está indicada para el tratamiento de la anemia asociada con hepatitis C.
A los pacientes con pérdida repentina de eficacia del tratamiento (manifestada por una disminución del nivel de hemoglobina de 1–2 g/dl por mes) y un aumento en la necesidad de transfusiones, deben derivarse para estudiar el recuento de reticulocitos y detectar las causas típicas de disminución de la respuesta clínica (deficiencia de hierro, ácido fólico, vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre, hemólisis o fibrosis de médula ósea de cualquier origen).
Ante una disminución paradójica de la hemoglobina y el desarrollo de anemia grave asociada con un bajo recuento de reticulocitos, debe interrumpirse inmediatamente el tratamiento con Hemax, determinar la presencia de anticuerpos contra la eritropoyetina y realizar un estudio de médula ósea para confirmar el diagnóstico de anemia eritroide verdadera (PRCA).
No se debe administrar tratamiento con otros agentes estimulantes de la eritropoyesis a estos pacientes, ya que existe la posibilidad de reacción cruzada.
Tratamiento de anemia sintomática en pacientes adultos y niños con insuficiencia renal crónica.
En pacientes con insuficiencia renal crónica que reciben epoetina alfa, debe controlarse regularmente el nivel de hemoglobina hasta alcanzar un nivel estable, y luego periódicamente. La tasa de aumento del nivel de hemoglobina debe ser de aproximadamente 1 g/dl (0,62 mmol/l) por mes y no debe exceder los 2 g/dl (1,25 mmol/l) por mes para minimizar el riesgo de hipertensión arterial.
En pacientes con insuficiencia renal crónica, el nivel alcanzado de hemoglobina no debe exceder el límite superior de la concentración deseada de hemoglobina en sangre. En estudios clínicos se observó un mayor riesgo de muerte y reacciones adversas graves cardiovasculares con el uso de agentes estimulantes de la eritropoyesis para alcanzar concentraciones de hemoglobina superiores a 12 g/dl (7,5 mmol/l).
Estudios clínicos controlados no demostraron ventajas significativas del uso de epoetinas con concentraciones de hemoglobina superiores al nivel necesario para controlar los síntomas de anemia y prevenir transfusiones.
Debe tenerse precaución al aumentar la dosis de Hemax en pacientes con insuficiencia renal crónica, ya que dosis acumuladas altas de eritropoyetina pueden asociarse con un mayor riesgo de mortalidad y trastornos cardiovasculares y cerebrovasculares graves. En pacientes con respuesta insuficiente al tratamiento con epoetinas, debe considerarse otras alternativas para superar dicha respuesta insuficiente.
El estado de los pacientes con insuficiencia renal crónica que reciben Hemax por vía subcutánea debe controlarse regularmente en busca de pérdida de eficacia del tratamiento, definida como disminución o pérdida de respuesta al tratamiento con epoetina alfa en pacientes que previamente respondieron al tratamiento. La pérdida de eficacia se caracteriza por una disminución sostenida del nivel de hemoglobina independientemente del aumento de la dosis de epoetina alfa.
Con regímenes de tratamiento con intervalos de dosificación prolongados (administración de epoetina alfa menos de una vez por semana), en algunos pacientes puede disminuir el nivel de hemoglobina, por lo que podrían necesitar un aumento de la dosis. Debe controlarse regularmente el nivel de hemoglobina.
En pacientes en hemodiálisis se han observado trombosis del shunt, especialmente en aquellos con tendencia a hipotensión o complicaciones de fístulas arteriovenosas (por ejemplo, estenosis, aneurismas, etc.). A estos pacientes se recomienda examinar el shunt y prevenir la trombosis mediante, por ejemplo, el uso de ácido acetilsalicílico.
En casos aislados se ha observado hiperkalemia, aunque no se ha establecido una relación causal. En pacientes con insuficiencia renal crónica debe controlarse el nivel de electrolitos en suero. En caso de aumento del nivel de potasio en sangre, además del tratamiento adecuado para la hiperkalemia, debe considerarse la posibilidad de suspender temporalmente Hemax hasta la normalización del nivel de potasio.
Debido al aumento del nivel de hematocrito, los pacientes en hemodiálisis que reciben Hemax a menudo requieren un aumento de la dosis de heparina durante la diálisis. Con una heparinización insuficiente, puede desarrollarse oclusión del sistema de diálisis.
Según la información disponible actualmente, el uso de Hemax para el tratamiento de anemia en adultos con insuficiencia renal pre-diálisis no acelera la progresión de la insuficiencia renal.
Tratamiento de pacientes con anemia inducida por quimioterapia.
En pacientes con enfermedades oncológicas que reciben epoetina alfa, debe controlarse regularmente el nivel de hemoglobina hasta alcanzar un nivel estable, y luego periódicamente.
Las epoetinas son factores de crecimiento que estimulan principalmente la producción de eritrocitos. Los receptores de eritropoyetina también se han detectado en la superficie de diversas células tumorales. Al igual que con otros factores de crecimiento, no puede excluirse la posibilidad de que las epoetinas estimulen el crecimiento de ciertos tipos de tumores.
No puede descartarse el impacto de los agentes estimulantes de la eritropoyesis sobre la progresión del tumor o la supervivencia sin progresión de la enfermedad. En estudios sobre el uso de epoetina alfa y otros agentes estimulantes de la eritropoyesis se asoció con una disminución del control locorregional del tumor o de la supervivencia general:
- disminución del control locorregional en pacientes con cáncer de cabeza y cuello en progresión que recibieron radioterapia, al usarlo con el fin de elevar el nivel de hemoglobina por encima de 14 g/dl (8,7 mmol/l);
- reducción de la supervivencia general y aumento de casos fatales debido a la progresión de la enfermedad durante 4 meses en pacientes con cáncer de mama metastásico que recibieron quimioterapia, al usarlo con el fin de elevar el nivel de hemoglobina a 12–14 g/dl (7,5–8,7 mmol/l);
- mayor riesgo de resultado fatal al usarlo con el fin de elevar el nivel de hemoglobina a 12 g/dl (7,5 mmol/l) en pacientes con enfermedad maligna activa que no reciben ni quimioterapia ni radioterapia. Los medicamentos estimulantes de la eritropoyesis están contraindicados en este grupo de pacientes;
- aumento del 9 % en el riesgo de progresión de la enfermedad o muerte en el grupo de pacientes que recibieron epoetina alfa y tratamiento estándar, y un aumento del 15 % en el riesgo, estadísticamente no excluible, en pacientes con cáncer de mama metastásico que recibieron quimioterapia, al usarlo con el fin de elevar el nivel de hemoglobina a 10–12 g/dl (6,2–7,5 mmol/l).
Debido a lo anterior, en algunas situaciones clínicas puede ser preferible la transfusión de sangre para tratar la anemia en pacientes con cáncer. La decisión sobre el uso de eritropoyetinas recombinantes debe basarse en una evaluación del beneficio-riesgo para cada paciente específico, considerando el contexto clínico particular. Los factores que deben considerarse en dicha evaluación incluyen el tipo y estadio del tumor; grado de anemia; esperanza de vida esperada; condiciones bajo las cuales se trata al paciente y los deseos del propio paciente.
En pacientes con enfermedades oncológicas que reciben quimioterapia, generalmente se observa un retraso de 2–3 semanas entre la administración de eritropoyetina y la aparición de glóbulos rojos inducidos por eritropoyetina. Esta característica debe tenerse en cuenta al evaluar la conveniencia del tratamiento (especialmente en pacientes con necesidad de transfusiones).
Pacientes sometidos a intervención quirúrgica y participantes en programas de recolección de sangre autóloga.
Debe seguirse todas las precauciones especiales relacionadas con el programa de recolección de sangre autóloga, especialmente los procedimientos para la recuperación del volumen circulante.
Pacientes antes de cirugía ortopédica electiva extensa.
Siempre deben seguirse las prácticas adecuadas de hemotransfusión durante el período pre y postoperatorio. Los pacientes antes de cirugía ortopédica electiva extensa deben recibir profilaxis antitrombótica adecuada, ya que pueden presentarse complicaciones trombóticas y vasculares tras la cirugía, especialmente en presencia de enfermedades cardiovasculares concomitantes. Debe tenerse especial precaución en el tratamiento de pacientes con predisposición al desarrollo de trombosis venosa profunda. Además, en pacientes con un nivel inicial de hemoglobina > 13 g/dl, la posibilidad de complicaciones trombóticas o vasculares postoperatorias asociadas con el tratamiento con epoetina alfa es significativamente mayor. Por lo tanto, no se recomienda el uso de epoetina alfa en pacientes con un nivel inicial de hemoglobina > 13 g/dl.
Dieta
Con el aumento del hematocrito, los pacientes experimentan un aumento del apetito, lo que conduce a un mayor consumo de alimentos. En este caso, deben tomarse medidas para prevenir el aumento del contenido de potasio en el organismo.
Propiedades carcinogénicas y mutagénicas
Las propiedades carcinogénicas de Hemax no han sido evaluadas. La epoetina no induce mutaciones genéticas en bacterias ni aberraciones cromosómicas en células de mamíferos.
Efecto sobre la fertilidad
En ratas preñadas se observó una tendencia a un ligero aumento en la frecuencia de muerte embrionaria tras la administración intravenosa de epoetina a dosis de 100–500 UI/kg.
Disminución de la eficacia del medicamento o ausencia de respuesta
Si en pacientes que reciben epoetina en dosis de mantenimiento se observa una disminución de la respuesta al medicamento o su ausencia total, deben descartarse las siguientes causas:
- deficiencia de hierro
- infección, inflamación, tumores
- pérdida oculta de sangre
- enfermedades hematológicas (talasemia, mielodisplasia, etc.)
- hemólisis
- intoxicación por aluminio
- deficiencia de vitamina B12 o ácido fólico
- fibrosis quística
- anemia eritroide verdadera.
Cloruro de sodio
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por dosis, es decir, es libre de sodio.
Uso durante el embarazo o la lactancia.
Embarazo
No se han realizado estudios suficientes sobre el uso de Hemax durante el embarazo. Los estudios en animales mostraron toxicidad reproductiva. Por lo tanto, este medicamento solo debe administrarse durante el embarazo si el beneficio potencial justifica el riesgo potencial para el feto. No se recomienda el uso de epoetina alfa en mujeres embarazadas que participan en programas de recolección de sangre autóloga.
En estudios con ratas preñadas se observó un ligero aumento en la frecuencia de muerte embrionaria, mientras que en conejas preñadas no se observaron efectos adversos tras la administración del medicamento a dosis de 500 UI/kg.
Lactancia.
La eritropoyetina humana está normalmente presente en la leche materna, aunque el papel de la eritropoyetina ingerida con la leche materna sigue sin aclararse. No se sabe si la epoetina alfa exógena se excreta en la leche materna. La epoetina alfa debe usarse con precaución en mujeres que amamantan. La decisión sobre continuar o interrumpir la lactancia o continuar o interrumpir el tratamiento con epoetina alfa debe tomarse considerando el beneficio de la lactancia para el niño y el beneficio del tratamiento con epoetina alfa para la mujer.
No se recomienda el uso de epoetina alfa en pacientes que participan en programas de recolección de sangre autóloga durante la lactancia.
Fertilidad.
No se han realizado estudios sobre el efecto de la epoetina alfa sobre la fertilidad en hombres o mujeres.
Capacidad para afectar la velocidad de reacción al conducir vehículos de motor u operar maquinaria.
No se han realizado estudios específicos al respecto, pero, considerando que durante el uso del medicamento Hemax pueden presentarse reacciones adversas como cefalea, fatiga y otras, las personas que toman este medicamento deben abstenerse de conducir vehículos de motor y operar maquinaria.
Vía de administración y dosis.
Todas las demás causas de anemia (deficiencia de hierro, ácido fólico, vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre, hemólisis o fibrosis de médula ósea de cualquier etiología) deben identificarse y tratarse antes de iniciar la terapia con epoetina alfa y, si es necesario, ajustar la dosis. Para lograr una respuesta óptima al tratamiento con epoetina alfa, debe asegurarse un aporte adecuado de hierro y, si es necesario, administrar suplementos de hierro (véase la sección «Precauciones de uso»).
Tratamiento de la anemia sintomática en pacientes adultos con insuficiencia renal crónica.
Los síntomas y complicaciones de la anemia pueden variar según la edad, sexo y enfermedades concomitantes; se requiere una evaluación individualizada por parte del médico del curso de la enfermedad y del estado del paciente.
El nivel deseado recomendado de hemoglobina es entre 10 g/dl y 12 g/dl (6,2–7,5 mmol/l). Hemax se utiliza con el fin de aumentar el nivel de hemoglobina hasta un máximo de 12 g/dl (7,5 mmol/l). Se debe evitar un aumento del nivel de hemoglobina superior a 2 g/dl (1,25 mmol/l) en 4 semanas. En tal caso, se debe reducir la dosis como se indica más adelante.
Debido a las características individuales, en algunos pacientes pueden observarse niveles de hemoglobina más altos o más bajos que el objetivo deseado. El nivel de hemoglobina debe controlarse mediante ajuste de la dosis, teniendo en cuenta el nivel recomendado entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l).
Se debe evitar un nivel constante de hemoglobina superior a 12 g/dl (7,5 mmol/l). Si la concentración de hemoglobina aumenta más de 2 g/dl (1,25 mmol/l) por mes o el nivel constante de hemoglobina supera los 12 g/dl (7,5 mmol/l), la dosis de Hemax debe reducirse en un 25 %. Si el nivel de hemoglobina supera los 13 g/dl (8,1 mmol/l), el tratamiento debe suspenderse hasta que el nivel de hemoglobina disminuya a 12 g/dl (7,5 mmol/l), y luego reanudarse con epoetina alfa a una dosis un 25 % menor que la anterior.
El estado del paciente debe vigilarse cuidadosamente para asegurar que se utiliza la dosis más baja eficaz de Hemax que controle la anemia y sus síntomas, manteniendo el nivel de hemoglobina no superior a 12 g/dl (7,5 mmol/l).
Debe tenerse precaución al aumentar las dosis de agentes estimulantes de la eritropoyesis en pacientes con insuficiencia renal crónica. En pacientes con mala respuesta al tratamiento con estos agentes, deben considerarse otras posibles causas de respuesta insatisfactoria (véase la sección «Precauciones de uso»).
El tratamiento con Hemax se divide en dos fases: fase de corrección y fase de mantenimiento.
Pacientes adultos en hemodiálisis.
Cuando se administra a pacientes en hemodiálisis, se prefiere la administración intravenosa.
Fase de corrección.
Dosis inicial: 50 UI/kg tres veces por semana.
Si es necesario, la dosis puede aumentarse o reducirse en 25 UI/kg (tres veces por semana) hasta alcanzar el nivel deseado de hemoglobina de 10–12 g/dl (6,2–7,5 mmol/l). Este ajuste de dosis debe realizarse progresivamente, con una frecuencia no mayor a una vez cada 4 semanas.
Fase de mantenimiento.
La dosis semanal recomendada oscila entre 75 y 300 UI/kg.
Para mantener el nivel deseado de hemoglobina entre 10 y 12 g/dl (6,2–7,5 mmol/l), se debe ajustar adecuadamente la dosis.
Los pacientes con un nivel inicial muy bajo de hemoglobina (< 6 g/dl, o < 3,75 mmol/l) pueden requerir una dosis de mantenimiento más alta que los pacientes con anemia menos grave al inicio del tratamiento (hemoglobina > 8 g/dl, o > 5 mmol/l).
Pacientes adultos con insuficiencia renal en período predialítico.
En pacientes con insuficiencia renal en período predialítico y sin catéter intravenoso establecido, el medicamento puede administrarse por vía subcutánea.
Fase de corrección.
Dosis inicial: 50 UI/kg tres veces por semana, aumentando si es necesario en 25 UI/kg (tres veces por semana), con intervalos entre aumentos de dosis de al menos 4 semanas, hasta alcanzar un nivel de hemoglobina entre 10–12 g/dl (6,2–7,5 mmol/l).
Fase de mantenimiento.
Durante la fase de mantenimiento, Hemax puede administrarse tres veces por semana o, en caso de administración subcutánea, una vez por semana o una vez cada dos semanas.
Las dosis y los intervalos entre administraciones deben ajustarse para mantener el nivel deseado de hemoglobina entre 10 y 12 g/dl (6,2–7,5 mmol/l). El alargamiento de los intervalos entre dosis puede requerir un aumento de la dosis.
La dosis máxima no debe exceder los 150 UI/kg tres veces por semana, 240 UI/kg (máximo hasta 20000 UI) una vez por semana o 480 UI/kg (máximo hasta 40000 UI) una vez cada dos semanas.
Pacientes adultos en diálisis peritoneal.
En ausencia de catéter intravenoso establecido, Hemax puede administrarse por vía subcutánea.
Fase de corrección.
Dosis inicial: 50 UI/kg dos veces por semana.
Fase de mantenimiento.
La dosis de mantenimiento recomendada es de 25 a 50 UI/kg dos veces por semana mediante la administración de dos inyecciones equivalentes.
La dosis debe ajustarse adecuadamente para mantener el nivel deseado de hemoglobina entre 10 y 12 g/dl (6,2–7,5 mmol/l).
Tratamiento de pacientes adultos con anemia inducida por quimioterapia.
Los síntomas y complicaciones de la anemia pueden variar según la edad, sexo y enfermedades concomitantes; se requiere una evaluación individualizada por parte del médico del curso de la enfermedad y del estado del paciente.
Hemax debe administrarse a pacientes con anemia (nivel de hemoglobina ≤ 10 g/dl (6,2 mmol/l)). Dosis inicial: 150 UI/kg subcutánea tres veces por semana.
Alternativamente, Hemax puede administrarse a una dosis de 450 UI/kg subcutánea una vez por semana.
La dosis debe ajustarse adecuadamente para mantener el nivel deseado de hemoglobina entre 10 y 12 g/dl (6,2–7,5 mmol/l). Se debe evitar un nivel constante de hemoglobina superior a 12 g/dl (7,5 mmol/l).
Debido a las características individuales, en algunos pacientes pueden observarse niveles de hemoglobina más altos o más bajos que el objetivo deseado. El nivel de hemoglobina debe controlarse mediante ajuste de la dosis, teniendo en cuenta el nivel recomendado entre 10 g/dl (6,2 mmol/l) y 12 g/dl (7,5 mmol/l).
Si tras 4 semanas de tratamiento el nivel de hemoglobina aumenta al menos 1 g/dl (0,6 mmol/l) o el recuento de reticulocitos aumenta ≥ 40000 células/µl, se debe continuar con Hemax a una dosis de 150 UI/kg tres veces por semana o 450 UI/kg una vez por semana.
Si tras 4 semanas de tratamiento con la dosis inicial el nivel de hemoglobina aumenta menos de 1 g/dl (0,62 mmol/l) o el nivel de reticulocitos aumenta menos de 40000 células/µl, la dosis debe aumentarse a 300 UI/kg tres veces por semana.
Si tras 4 semanas adicionales de tratamiento con 300 UI/kg tres veces por semana el nivel de hemoglobina aumenta al menos 1 g/dl (0,62 mmol/l) o el recuento de reticulocitos aumenta al menos 40000 células/µl, se debe continuar con Hemax a 300 UI/kg tres veces por semana.
Sin embargo, si el nivel de hemoglobina aumenta menos de 1 g/dl (menos de 0,62 mmol/l) o el nivel de reticulocitos aumenta menos de 40000 células/µl respecto al nivel inicial, la respuesta clínica se considera dudosa y el tratamiento debe interrumpirse.
Ajuste de la dosis para mantener el nivel objetivo de hemoglobina entre 10–12 g/dl.
Si el nivel de hemoglobina aumenta más de 2 g/dl (1,25 mmol/l) en un mes o si supera los 12 g/dl (7,5 mmol/l), se debe reducir la dosis de Hemax en un 25–50 %. Si el nivel de hemoglobina supera los 13 g/dl (8,1 mmol/l), el tratamiento debe suspenderse temporalmente hasta que el nivel descienda por debajo de 12 g/dl (7,5 mmol/l), y luego reanudarse con una dosis un 25 % menor que la anterior.
Esquema del régimen de dosificación recomendado:
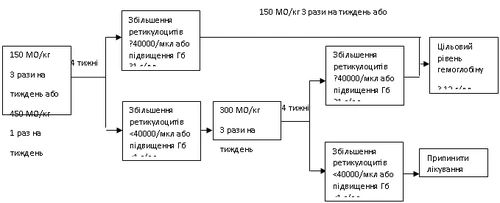
El estado del paciente debe vigilarse cuidadosamente para asegurar que se utiliza la dosis más baja aprobada de Hemax para controlar los síntomas de la anemia.
El uso de Hemax debe continuar durante un mes tras la finalización de la quimioterapia.
Pacientes adultos participantes en un programa de donación autóloga de sangre antes de intervenciones quirúrgicas.
A los pacientes con anemia moderada (hematocrito entre 33–39 %) que requieran ≥ 4 unidades de sangre, se debe tratar con Hemax a una dosis de 600 UI/kg intravenosa dos veces por semana durante 3 semanas antes de la intervención quirúrgica. Hemax se administra tras cada procedimiento de extracción de sangre.
Pacientes adultos antes de una intervención ortopédica planificada de gran envergadura.
La dosis recomendada es 600 UI/kg subcutánea una vez por semana durante 3 semanas previas a la cirugía (días 21, 14 y 7 antes de la operación) y el día de la cirugía.
Si por indicaciones médicas es necesario acortar el período preoperatorio a menos de 3 semanas, Hemax debe administrarse diariamente a una dosis de 300 UI/kg subcutánea durante 10 días consecutivos antes de la cirugía, el día de la cirugía y durante 4 días posteriores. Si durante el período preoperatorio el nivel de hemoglobina alcanza 15 g/dl o más, el uso de Hemax debe suspenderse completamente y no deben administrarse las dosis siguientes.
Pacientes adultos con SMD de riesgo bajo o intermedio-1.
Hemax debe administrarse a pacientes con anemia sintomática (nivel de hemoglobina ≤ 10 g/dl (6,2 mmol/l)).
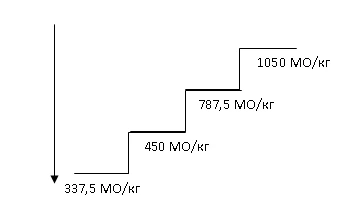
La dosis inicial recomendada de Hemax es 450 UI/kg (dosis total máxima: 40000 UI) subcutánea una vez por semana, con un intervalo mínimo de 5 días entre dosis.
La dosis debe ajustarse adecuadamente para mantener el nivel deseado de hemoglobina entre 10 y 12 g/dl (6,2–7,5 mmol/l). Debe evaluarse la respuesta al tratamiento con Hemax entre las 8 y 12 semanas tras el inicio del tratamiento. Los aumentos y reducciones de dosis deben realizarse de forma escalonada (véase el diagrama a continuación). Se debe evitar un nivel de hemoglobina superior a 12 g/dl (7,5 mmol/l).
Aumento de la dosis. La dosis no debe aumentarse más de 1050 UI/kg (dosis total: 80000 UI) por semana. Si se pierde la respuesta al tratamiento o si la concentración de hemoglobina disminuye ≥ 1 g/dl tras una reducción de dosis, se debe aumentar la dosis un escalón. Entre aumentos de dosis debe transcurrir al menos 4 semanas.
Mantenimiento y reducción de la dosis. El tratamiento con Hemax debe suspenderse si la concentración de hemoglobina supera los 12 g/dl (7,5 mmol/l). Tan pronto como el nivel de hemoglobina sea < 11 g/dl, debe reanudarse la administración con la misma dosis o una dosis un escalón más baja, según decisión médica. Debe considerarse la reducción de la dosis si se observa un rápido aumento del nivel de hemoglobina (> 2 g/dl en 4 semanas).
 |
|||||
  |
|||||
Los síntomas y complicaciones de la anemia pueden variar dependiendo de la edad, el sexo y las enfermedades concomitantes; es necesario evaluar individualmente el curso de la enfermedad y el estado del paciente.
Niños.
Tratamiento de la anemia sintomática en niños con insuficiencia renal crónica sometidos a hemodiálisis.
Los síntomas y complicaciones de la anemia pueden variar dependiendo de la edad, el sexo y las enfermedades concomitantes; es necesario evaluar individualmente el curso de la enfermedad y el estado del paciente.
En niños, el nivel recomendado de hemoglobina oscila entre 9,5 g/dl y 11 g/dl (5,9–6,8 mmol/l). HEMAX debe utilizarse para aumentar el nivel de hemoglobina hasta un máximo de 11 g/dl (6,8 mmol/l). Debe evitarse un aumento del nivel de hemoglobina superior a 2 g/dl (1,25 mmol/l) en 4 semanas.
En tal caso, la dosis debe ajustarse adecuadamente.
El estado del paciente debe vigilarse cuidadosamente para asegurar que se utiliza la dosis más baja aprobada de HEMAX necesaria para controlar los síntomas de la anemia.
El tratamiento se divide en dos fases: fase de corrección y fase de mantenimiento.
Cuando se dispone de un catéter intravenoso establecido, se prefiere la vía intravenosa.
Fase de corrección.
Dosis inicial: 50 UI/kg por vía intravenosa, 3 veces por semana.
Si es necesario, la dosis puede reducirse o aumentarse en 25 UI/kg (3 veces por semana) hasta alcanzar el nivel deseado de hemoglobina entre 9,5 g/dl y 11 g/dl (5,9–6,8 mmol/l). Este ajuste de la dosis debe realizarse progresivamente, no con más frecuencia de una vez cada 4 semanas.
Fase de mantenimiento.
La dosis debe ajustarse adecuadamente con el fin de mantener el nivel óptimo de hemoglobina entre 9,5 g/dl y 11 g/dl (5,9–6,8 mmol/l).
En general, los niños con un peso corporal inferior a 30 kg requieren una dosis de mantenimiento mayor que los adultos y los niños con un peso corporal superior a 30 kg. Los pacientes pediátricos con un nivel inicial muy bajo de hemoglobina (< 6,8 g/dl o < 4,25 mmol/l) pueden requerir dosis de mantenimiento más altas en comparación con aquellos pacientes cuyo nivel inicial de hemoglobina fue más elevado (> 6,8 g/dl o > 4,25 mmol/l).
Tratamiento de la anemia en niños con insuficiencia renal crónica en período predialítico o sometidos a diálisis peritoneal.
La seguridad y eficacia del uso de HEMAX en niños con insuficiencia renal crónica y anemia que aún no están en diálisis o que están en diálisis peritoneal no han sido establecidas. Actualmente no existen recomendaciones de dosificación para este grupo de pacientes.
Tratamiento de niños con anemia inducida por quimioterapia.
La seguridad y eficacia del uso de HEMAX en niños con anemia inducida por quimioterapia no han sido establecidas.
Tratamiento de niños que participan en programas de donación autóloga de sangre antes de intervenciones quirúrgicas.
La seguridad y eficacia del uso de HEMAX en niños que participan en programas de donación autóloga de sangre antes de grandes intervenciones quirúrgicas no han sido establecidas.
Tratamiento de niños antes de una intervención ortopédica quirúrgica programada.
La seguridad y eficacia del uso de HEMAX en niños sometidos a una intervención ortopédica quirúrgica programada no han sido establecidas.
Vía de administración
La epoetina alfa puede administrarse mediante inyecciones subcutáneas e intravenosas.
Procedimiento para la preparación de la solución inyectable: añadir a cada frasco con el liofilizado la cantidad de agua para inyección indicada en la tabla siguiente.
| Ingrediente activo |
1000 UMI |
2000 UMI |
3000 UMI |
4000 UMI |
10 000 UMI |
20 000 UMI |
40 000 UMI |
| Agua para inyección |
1 ml |
2 ml |
2 ml |
2 ml |
1 ml |
1 ml |
1 ml |
Como con cualquier medicamento administrado por vía parenteral, el epoetina alfa debe examinarse antes de su uso para verificar la ausencia de partículas extrañas visibles y cualquier cambio en el color de la solución.
Administración intravenosa.
A los pacientes adultos que participan en programas de donación autóloga de sangre previa a intervenciones quirúrgicas, se les administra Hemax por vía intravenosa. En el tratamiento de anemia sintomática en adultos y niños con insuficiencia renal crónica cuando existe acceso intravenoso (pacientes en hemodiálisis), se prefiere la administración por vía intravenosa.
Hemax se administra mediante inyección intravenosa lenta de 1 a 5 minutos, según la dosis del medicamento. En pacientes sometidos a hemodiálisis, la inyección en bolo puede administrarse directamente durante el procedimiento a través del puerto venoso adecuado en la línea de diálisis. Alternativamente, el medicamento puede administrarse tras finalizar la sesión de hemodiálisis a través de la fístula del catéter, seguido de una inyección de 10 ml de solución de cloruro sódico isotónica para lavar el sistema y asegurar una adecuada distribución del fármaco en la circulación.
La administración lenta es recomendable en pacientes que presenten síntomas similares a los de la gripe durante el tratamiento (ver sección «Reacciones adversas»).
Hemax no debe administrarse mediante infusión intravenosa ni mezclarse con otros medicamentos.
Administración subcutánea.
En adultos con anemia inducida por quimioterapia, en pacientes adultos antes de una intervención quirúrgica ortopédica mayor programada, y en adultos con síndrome mielodisplásico de riesgo bajo o intermedio-1, Hemax se administra por vía subcutánea.
En el tratamiento de anemia sintomática en adultos con insuficiencia renal crónica en período predialítico o en diálisis peritoneal, cuando no existe un catéter intravenoso colocado, puede administrarse Hemax por vía subcutánea.
El volumen máximo de administración subcutánea del medicamento en un solo sitio es de 1 ml. Si se requiere un volumen mayor, la inyección subcutánea debe realizarse en varios sitios.
El medicamento debe administrarse subcutáneamente en las extremidades o en la pared anterior del abdomen.
Si el médico considera que el paciente o el cuidador puede administrar Hemax subcutáneamente de forma segura y eficaz, se les debe proporcionar instrucciones adecuadas sobre la dosificación y el uso correcto.
Niños.
La epoetina alfa está indicada para el tratamiento de la anemia asociada a la insuficiencia renal crónica en niños de 1 a 18 años de edad que estén en diálisis. No se ha establecido la seguridad y eficacia del medicamento en niños menores de 1 mes de edad.
Sobredosis.
El medicamento tiene un amplio margen terapéutico. En caso de sobredosis de epoetina alfa, pueden presentarse efectos que reflejan el grado máximo de acción farmacológica de la hormona, es decir, policitemia y síntomas asociados como cefalea, mareo, somnolencia, etc. En caso de niveles excesivamente elevados de hemoglobina, puede considerarse la flebotomía. Si es necesario, se aplicará tratamiento sintomático.
En caso de sobredosis, debe acudirse inmediatamente al centro médico más cercano o al centro de toxicología.
Reacciones adversas.
La reacción adversa no deseada más frecuente durante el tratamiento con epoetina alfa es el aumento dependiente de la dosis de la presión arterial o el empeoramiento de la hipertensión preexistente. Es necesario controlar la presión arterial desde el inicio del tratamiento. Otras reacciones adversas frecuentes observadas durante estudios clínicos con epoetina alfa incluyen trombosis venosa profunda, embolia pulmonar, convulsiones, diarrea, náuseas, cefalea, estado gripal, pirexia, erupciones cutáneas y vómitos.
Principalmente al comienzo del tratamiento, pueden presentarse síntomas similares a los del resfriado, como cefalea, dolor muscular y articular, y escalofríos. La frecuencia puede variar según la indicación.
Durante estudios sobre el uso del medicamento con intervalos entre dosis prolongados en pacientes adultos con insuficiencia renal en período prehemodiálisis, se observó deterioro de la permeabilidad de las vías respiratorias, incluyendo las vías respiratorias superiores, congestión nasal y nasofaringitis.
En pacientes que recibieron tratamiento con agentes estimulantes de la eritropoyesis, se ha observado una mayor frecuencia de complicaciones trombovasculares (véase la sección «Propiedades farmacodinámicas»).
Albúmina (humana)
Hemax contiene albúmina obtenida de sangre humana. El riesgo de transmisión de infecciones virales es extremadamente bajo debido a la tecnología empleada en la obtención de este componente. Asimismo, el riesgo de transmisión del agente causal de la enfermedad de Creutzfeldt-Jakob es extremadamente improbable. No se han descrito casos de transmisión de agentes infecciosos víricos mediante albúmina.
Frecuencia de aparición de reacciones adversas: muy frecuente (≥1/10); frecuente (≥1/100 hasta <1/10); poco frecuente (≥1/1000 hasta <1/100); rara (≥1/10000 hasta <1/1000); muy rara (<1/10000); frecuencia desconocida (no puede determinarse con los datos disponibles).
Trastornos de la sangre y del sistema linfático.
Rara: trombocitemia, aplasia eritroide verdadera.
Sistema inmunitario.
Poco frecuente: reacciones de hipersensibilidad.
Rara: reacciones anafilácticas: complicaciones potencialmente graves asociadas con alteraciones respiratorias o con disminución de la TA; reacciones inmunitarias (tiene mínima capacidad para inducir la formación de anticuerpos).
Sistema nervioso.
Frecuente: cefalea.
Poco frecuente: convulsiones, accidente cerebrovascular, hemorragia cerebral.
Frecuencia desconocida: accidente cerebrovascular, encefalopatía hipertensiva, ataque isquémico transitorio, vértigo, somnolencia.
Órganos de la vista.
Frecuencia desconocida: trombosis de venas de la retina.
Corazón.
Frecuencia desconocida: infarto de miocardio.
Sistema vascular.
Frecuente: trombosis venosas y arteriales, hipertensión arterial.
Frecuencia desconocida: trombosis venosa profunda (en pacientes con insuficiencia renal crónica), crisis hipertensiva.
Sistema respiratorio, órganos torácicos y mediastino.
Frecuente: embolia pulmonar (en pacientes con cáncer), tos.
Poco frecuente: deterioro de la permeabilidad de las vías respiratorias.
Frecuencia desconocida: embolia pulmonar (en pacientes con insuficiencia renal crónica).
Aparato gastrointestinal.
Muy frecuente: náuseas, diarrea, vómitos.
Piel y tejido subcutáneo.
Frecuente: erupciones cutáneas, eccema.
Poco frecuente: urticaria.
Frecuencia desconocida: angioedema, prurito, edema de Quincke, síndrome de Stevens-Johnson, necrólisis epidérmica tóxica (pueden poner en peligro la vida o tener consecuencias letales).
Músculo esquelético, tejido conjuntivo y huesos.
Frecuente: artralgia, dolor óseo, dolor en extremidades, mialgia.
Trastornos congénitos, hereditarios/genéticos.
Rara: porfiria aguda.
Trastornos generales y condiciones en el sitio de administración.
Muy frecuente: fiebre, pirexia (en pacientes con cáncer).
Frecuente: estado gripal, escalofríos, reacciones en el sitio de inyección, edema periférico.
Frecuencia desconocida: escalofríos, falta de respuesta al tratamiento.
Pruebas de laboratorio.
Rara: presencia de anticuerpos contra eritropoyetina.
Frecuencia desconocida: hipofosfatemia, aumento de la concentración de urea, creatinina y ácido úrico en plasma sanguíneo (en pacientes con insuficiencia renal crónica).
Metabolismo y nutrición.
Poco frecuente: hiperkalemia (frecuente en pacientes sometidos a hemodiálisis).
Lesiones, intoxicaciones y complicaciones procedimentales.
Frecuente: trombosis del shunt, incluyendo el equipo para diálisis (en pacientes con insuficiencia renal crónica).
Pacientes con insuficiencia renal crónica.
En pacientes con insuficiencia renal crónica, un nivel de hemoglobina superior a 12 g/dL puede asociarse con un mayor riesgo de complicaciones cardiovasculares, incluyendo consecuencias letales.
En pacientes sometidos a hemodiálisis, especialmente en caso de predisposición a hipotensión o presencia de complicaciones en la fístula arteriovenosa (estenosis, aneurismas, etc.), se han descrito casos de trombosis del shunt.
Pacientes con enfermedades oncológicas.
El desarrollo de complicaciones trombóticas es posible en pacientes que reciben terapia con agentes estimulantes de la eritropoyesis, incluyendo la epoetina alfa.
Pacientes quirúrgicos adultos.
No puede descartarse la posibilidad de que el tratamiento con epoetina alfa en pacientes con niveles estables de hemoglobina >13 g/dL pueda asociarse con un mayor riesgo de complicaciones trombóticas/vasculares postoperatorias.
Descripción de reacciones adversas individuales
Trombosis venosas y arteriales, con y sin consecuencias letales, tales como trombosis venosa profunda, embolia pulmonar, trombosis retiniana, trombosis arterial (incluyendo infarto de miocardio e isquemia miocárdica), trombosis de la retina y trombosis del shunt (incluyendo oclusión del sistema de diálisis), trombosis en la zona del anastomosis arteriovenoso. También pueden observarse complicaciones cerebrovasculares (incluyendo accidente cerebrovascular isquémico, hemorragias cerebrales) y ataques isquémicos transitorios, aneurismas.
Se han notificado reacciones de hipersensibilidad, particularmente con erupciones cutáneas (incluyendo urticaria), reacciones anafilácticas y angioedema.
Se han notificado casos graves de reacciones adversas cutáneas relacionadas con el tratamiento con epoetinas, incluyendo el síndrome de Stevens-Johnson y la necrólisis epidérmica tóxica, que pueden poner en peligro la vida o tener consecuencias letales (véase la sección «Propiedades farmacodinámicas»).
Se han observado casos de crisis hipertensiva con encefalopatía y convulsiones, que requirieron evaluación médica inmediata y tratamiento intensivo, en pacientes con presión arterial normal o baja al inicio del tratamiento. Debe prestarse especial atención a la aparición de cefalea súbita, punzante y tipo migraña, que puede ser un signo de alarma.
Aplasia eritroide verdadera
Dado que la epoetina alfa es un medicamento proteico, en algunos pacientes puede producirse formación de anticuerpos contra ella. Casos raros de aplasia eritroide verdadera en pacientes con insuficiencia renal que reciben el medicamento por vía subcutánea están asociados con la presencia de anticuerpos neutralizantes contra la epoetina alfa. En tales casos, el uso de cualquier medicamento que contenga epoetina está contraindicado.
Muy rara (<1/10000 casos por paciente-año) se han notificado casos de aplasia eritroide verdadera mediada por anticuerpos (PRCA) en pacientes que recibieron tratamientos con eritropoyetina durante meses o años.
Pacientes adultos con SDM de riesgo bajo o intermedio-1
En el transcurso de un estudio clínico, se produjeron eventos trombovasculares en 4 (4,7%) pacientes (muerte súbita, accidente cerebrovascular isquémico, embolia y flebitis). Todos los eventos trombovasculares ocurrieron en el grupo tratado con epoetina alfa y durante las primeras 24 semanas de tratamiento. Tres casos fueron confirmados; el cuarto caso (muerte súbita) no fue confirmado. Dos pacientes presentaban factores de riesgo importantes (fibrilación auricular, insuficiencia cardíaca y tromboflebitis).
Niños con insuficiencia renal crónica en hemodiálisis.
La experiencia con el uso de eritropoyetina en niños con insuficiencia renal crónica en hemodiálisis, tanto durante estudios clínicos como en el período poscomercialización, es limitada. No se han identificado reacciones adversas específicas de la edad pediátrica, ni reacciones adversas que no correspondan a la enfermedad subyacente.
La notificación de reacciones adversas tras la comercialización del medicamento es importante. Permite realizar un seguimiento continuo de la relación beneficio/riesgo del medicamento. Se recomienda a los profesionales sanitarios notificar cualquier posible reacción adversa a través del sistema nacional de notificación.
Duración del efecto terapéutico. 2 años.
Condiciones de almacenamiento.
Conservar en lugares inaccesibles para los niños, a una temperatura no superior a 25 °C.
Envase.
1 frasco por caja de cartón.
Categoría de dispensación. Bajo receta médica.
Fabricante. Biosidus S.A.
Biosidus S.A.
Domicilio del fabricante y dirección del lugar de actividad.
Constitución 4234 (código postal C1254ABX), Ciudad de Buenos Aires, República Argentina
Constitución 4234 (código postal C1254ABX), Ciudad de Buenos Aires, República Argentina.