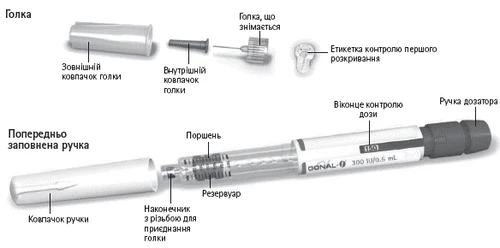
Gonal-f®
Ucrania
Contenido
INSTRUCCIONES para uso médico del medicamento GONAL-fâ (GONAL-fâ)
Composición:
Principio activo: fólitropina alfa (hormona folículoestimulante humana recombinante – rhFSH);
Un bolígrafo destinado a la administración:
300 UI de fólitropina alfa, equivalentes a 22 mcg, en 0,48 ml de solución; o
450 UI de fólitropina alfa, equivalentes a 33 mcg, en 0,72 ml de solución; o
900 UI de fólitropina alfa, equivalentes a 66 mcg, en 1,44 ml de solución;
Excipientes: poloxámero 188, sacarosa, metionina, fosfato de sodio dihidrato, fosfato sódico monobásico monohidrato, m-cresol, ácido fosfórico concentrado, hidróxido de sodio, agua para inyección.
Forma farmacéutica. Solución inyectable.
Propiedades físico-químicas principales: solución, prácticamente libre de partículas visibles.
Grupo farmacoterapéutico. Hormonas sexuales y moduladores del sistema sexual. Gonadotropinas.
Código ATC G03G A05.
Propiedades farmacológicas.
Farmacodinámica.
GONAL-f® es un preparado de la hormona estimulante del folículo (FSH) obtenida mediante técnicas de ingeniería genética a partir de células del ovario del hámster chino.
La hormona estimulante del folículo (FSH) y la hormona luteinizante (LH) son segregadas por la porción anterior de la hipófisis en respuesta a la hormona liberadora de gonadotropinas (GnRH) y desempeñan un papel complementario en los procesos de desarrollo folicular y ovulación. La FSH estimula el desarrollo de los folículos ováricos, mientras que la LH participa en los procesos de desarrollo folicular, esteroidogénesis y maduración.
Tras la administración de FSH recombinante (r-hFSH), se produce un aumento de los niveles de inhibina y estradiol (E2), seguido de la inducción del desarrollo folicular. Los niveles séricos de inhibina aumentan rápidamente y pueden detectarse ya desde el tercer día tras la administración de r-hFSH, mientras que el incremento de los niveles de E2 requiere más tiempo y generalmente se observa a partir del cuarto día del tratamiento. El volumen folicular total comienza a aumentar tras 4-5 días de administración diaria de r-hFSH, y dependiendo de la respuesta de la paciente, el efecto máximo se alcanza aproximadamente a los 10 días desde el inicio del tratamiento con r-hFSH.
En estudios clínicos, las pacientes con deficiencia grave de FSH y LH se definieron por niveles séricos de LH endógena < 1,2 UI/l; sin embargo, debe tenerse en cuenta que los resultados de las mediciones de LH pueden variar entre diferentes laboratorios.
En estudios clínicos comparativos, durante procedimientos de tecnologías de reproducción asistida (TRA) y en la inducción de la ovulación, GONAL-f® demostró mayor eficacia que la FSH urinaria, lo que se reflejó en una dosis total más baja y un período de tratamiento más corto necesarios para inducir la maduración folicular. En el contexto de las TRA, el uso de una dosis total más baja de GONAL-f® durante un período más corto permitió obtener un mayor número de ovocitos y embriones en división al segundo día tras la fecundación, en comparación con la FSH urinaria. En la inducción de la ovulación, el uso de GONAL-f® permitió lograr una menor frecuencia de cancelación de ciclos debido a la ineficacia del tratamiento, en comparación con la FSH urinaria.
Resultados del estudio GF 8407: estudio aleatorizado con diseño de grupos paralelos, en el que se compararon la eficacia y la seguridad del uso de GONAL-f® y de la FSH urinaria en el marco de TRA.
| Indicador |
ГОНАЛ-ф® (n = 130) |
FSH urinario (n = 116) |
| Número de ovocitos obtenidos |
11,0 ± 5,9 |
8,8 ± 4,8 |
| Duración de la estimulación, días |
11,7 ± 1,9 |
14,5 ± 3,3 |
| Dosis total necesaria de FSH (número de viales de 75 UI de FSH) |
27,6 ± 10,2 |
40,7 ± 13,6 |
| Necesidad de aumento de dosis (%) |
56,2 |
85,3 |
La diferencia entre los dos grupos fue estadísticamente significativa (p < 0,05) en todos los criterios mencionados.
La administración conjunta de GONAL-fâ y gonadotropina coriónica humana (hCG) durante al menos 4 meses induce la espermatogénesis en hombres con deficiencia de FSH.
Farmacocinética.
No se produce interacción farmacocinética cuando se administran simultáneamente folitropina alfa y lutropina alfa.
Distribución
Después de la administración intravenosa, la folitropina alfa se distribuye al líquido intersticial con una semivida inicial de aproximadamente 2 horas, y se elimina del organismo con una semivida terminal de entre 14 y 17 horas. El volumen de distribución en estado de equilibrio oscila entre 9 y 11 l.
Después de la administración subcutánea, la biodisponibilidad absoluta es del 66 % y la semivida terminal aparente se sitúa entre 24 y 59 horas. Tras la administración subcutánea, se ha demostrado una relación proporcional entre los parámetros farmacocinéticos y la dosis en un rango de dosis de hasta 900 UI. La administración repetida de folitropina alfa provoca un aumento de su acumulación hasta triplicarla, alcanzando un estado de equilibrio en 3-4 días.
Eliminación
El aclaramiento total es de 0,6 l/h; aproximadamente el 12 % de la dosis de folitropina alfa se excreta en la orina.
Características clínicas.
Indicaciones.
Tratamiento en mujeres adultas
- Anovulación (incluyendo el síndrome de ovario poliquístico) en mujeres que han demostrado ser resistentes al tratamiento con citrato de clomifeno.
- Estimulación del desarrollo folicular múltiple en pacientes sometidas a superovulación como parte de técnicas de reproducción asistida (ART), tales como fertilización in vitro (FIV), transferencia de gametos a las trompas de Falopio (GIFT) y transferencia de cigoto a las trompas de Falopio (ZIFT).
- Estimulación del desarrollo folicular en mujeres con deficiencia grave de hormona luteinizante (LH) y hormona foliculoestimulante (FSH), en combinación con un preparado que contenga LH.
Tratamiento en hombres adultos
- Estimulación de la espermatogénesis en hombres con hipogonadismo hipogonadotrópico congénito o adquirido, junto con tratamiento con gonadotropina coriónica humana (hCG).
Contraindicaciones.
- Hipersensibilidad al principio activo o a cualquier excipiente del medicamento;
- tumores del hipotálamo o de la hipófisis;
- aumento del tamaño de los ovarios o quistes no relacionados con el síndrome de ovario poliquístico o de origen desconocido;
- hemorragias ginecológicas de origen desconocido;
- carcinoma de ovario, útero o mama.
GONAL-fâ no debe administrarse en casos en los que no se pueda obtener una respuesta terapéutica eficaz, por ejemplo en:
- insuficiencia ovárica primaria;
- malformaciones congénitas de los órganos genitales incompatibles con el embarazo;
- fibromas uterinos incompatibles con el embarazo;
- insuficiencia testicular primaria.
Interacción con otros medicamentos y otras formas de interacción.
La administración concomitante de GONAL-fâ con otros medicamentos utilizados para estimular la ovulación (como hCG o citrato de clomifeno) puede potenciar la respuesta folicular, mientras que la administración conjunta con agonistas o antagonistas de la hormona liberadora de gonadotropinas (GnRH), que inducen la desensibilización de la hipófisis, puede requerir un aumento de la dosis de GONAL-fâ necesaria para lograr una respuesta ovárica adecuada. No se han notificado otras interacciones medicamentosas clínicamente relevantes durante el tratamiento con GONAL-fâ.
Características de aplicación.
Seguimiento
Para mejorar el seguimiento de los medicamentos biológicos, se debe registrar claramente el nombre y el número de lote del medicamento administrado.
Recomendaciones generales
Dado que GONAL-f* posee una marcada actividad gonadotrópica capaz de provocar reacciones adversas de intensidad leve a grave, solo médicos bien familiarizados con los problemas de infertilidad y sus métodos de tratamiento deben prescribir este medicamento.
La terapia con gonadotropinas requiere una dedicación temporal por parte del médico y otros profesionales sanitarios, así como el uso de equipos adecuados para el seguimiento del tratamiento. El uso seguro y eficaz de GONAL-f* en mujeres requiere un monitoreo regular de la respuesta ovárica mediante ecografía, preferiblemente combinado con la determinación de los niveles séricos de estradiol. La respuesta de los pacientes a la administración de FSH es individual: algunos pacientes responden muy débilmente a la FSH, mientras que otros lo hacen en exceso. Para el tratamiento tanto de mujeres como de hombres, se debe utilizar la dosis más baja eficaz según el objetivo terapéutico.
Pacientes con porfiria
Los pacientes con porfiria o antecedentes familiares de porfiria deben estar bajo estricta supervisión médica durante el tratamiento con GONAL-f*. En caso de aparición de los primeros signos de esta condición o empeoramiento de la misma, puede ser necesario interrumpir el tratamiento.
Tratamiento de mujeres
Antes de iniciar el tratamiento, la pareja infértil debe someterse a un examen para detectar contraindicaciones existentes o probables para el embarazo. En particular, los pacientes deben evaluarse para detectar hipotiroidismo, insuficiencia suprarrenal, hiperprolactinemia y recibir el tratamiento específico adecuado.
Durante la estimulación del crecimiento folicular, ya sea en el tratamiento de la infertilidad anovulatoria o en procedimientos de reproducción asistida (ART), las pacientes pueden presentar agrandamiento ovárico o desarrollar hiperestimulación ovárica. El cumplimiento de la dosis recomendada y del régimen de administración de GONAL-f*, así como un seguimiento cuidadoso del tratamiento, reducirán la frecuencia de estos eventos. La interpretación precisa de los indicadores de desarrollo y maduración folicular requiere la intervención de un especialista con experiencia en la interpretación de las pruebas correspondientes.
En estudios clínicos se ha demostrado un aumento de la sensibilidad ovárica al efecto de GONAL-f* cuando se administra conlutropina alfa simultáneamente. Si se considera necesario aumentar la dosis de FSH, es preferible hacerlo en intervalos de 7-14 días, incrementándola en 37,5-75 UI. No se ha realizado una comparación directa entre el uso de GONAL-f*/LH y gonadotropina menopáusica humana (hMG). La comparación con datos publicados sugiere que la frecuencia de ovulación obtenida con GONAL-f*/LH es similar a la obtenida con hMG.
Síndrome de hiperestimulación ovárica (OHSS)
Un resultado esperado de la estimulación ovárica controlada es un cierto aumento del tamaño de los ovarios. Este fenómeno, más frecuente en mujeres con síndrome de ovario poliquístico, generalmente desaparece sin tratamiento específico.
A diferencia del agrandamiento ovárico no complicado, el OHSS es un síndrome que progresa en gravedad. Incluye un aumento notable del tamaño de los ovarios, niveles séricos elevados de esteroides sexuales y un aumento de la permeabilidad vascular que puede provocar acumulación de líquido en la cavidad abdominal, pleural y, rara vez, pericárdica.
En casos graves de OHSS, pueden presentarse síntomas como dolor y sensación de distensión abdominal, aumento significativo del tamaño de los ovarios, aumento de peso, disnea, oliguria y síntomas gastrointestinales como náuseas, vómitos y diarrea. En el examen clínico pueden detectarse hipovolemia, hemoconcentración, desequilibrio electrolítico, ascitis, hemoperitoneo, derrames pleurales, hidrotórax o síndrome de distrés respiratorio agudo. En casos muy aislados, el OHSS grave puede complicarse con torsión ovárica y eventos tromboembólicos como embolia de la arteria pulmonar, accidente cerebrovascular isquémico e infarto de miocardio.
Los factores de riesgo independientes para el desarrollo de OHSS incluyen edad joven, bajo peso corporal, síndrome de ovario poliquístico, dosis altas de gonadotropinas exógenas, niveles séricos elevados o rápidamente crecientes de estradiol, episodios previos de OHSS, gran número de folículos ováricos en crecimiento o gran número de ovocitos recuperados en ciclos de técnicas de reproducción asistida (ART).
El cumplimiento de la dosis recomendada y del régimen de administración de GONAL-f* puede minimizar el riesgo de hiperestimulación ovárica. Se recomienda el monitoreo de los ciclos de estimulación mediante ecografía y determinación de estradiol para detectar precozmente los factores de riesgo.
Hay evidencia que sugiere que la hCG desempeña un papel clave en la iniciación del OHSS y que este síndrome puede empeorar y prolongarse si se produce un embarazo. Por lo tanto, ante signos de hiperestimulación ovárica, se recomienda suspender la administración de hCG y aconsejar a la paciente abstenerse de relaciones sexuales o usar métodos de barrera anticonceptivos durante al menos 4 días. El OHSS puede progresar rápidamente (dentro de las 24 horas) y convertirse en una complicación médica grave en pocos días. Generalmente ocurre tras la interrupción del tratamiento hormonal y alcanza su máxima frecuencia aproximadamente entre los días 7 y 10 después de finalizar el tratamiento. Por ello, tras la administración de hCG, la paciente debe permanecer bajo supervisión médica durante al menos 2 semanas.
En procedimientos de ART, la frecuencia de hiperestimulación puede reducirse mediante la aspiración de todos los folículos antes de la ovulación.
Generalmente, las formas leves o moderadas de OHSS desaparecen espontáneamente. Si se presenta una forma grave de OHSS, debe interrumpirse el tratamiento con gonadotropinas (si aún continúa), hospitalizar a la paciente y comenzar el tratamiento específico para OHSS.
Embarazo múltiple
En pacientes sometidas a inducción de ovulación, la frecuencia de embarazos múltiples es mayor que en la fecundación natural. La mayoría de los embarazos múltiples son gemelares. El embarazo múltiple, especialmente de orden superior, conlleva un mayor riesgo de resultados adversos en el parto y el período perinatal.
Para minimizar el riesgo de embarazo múltiple, se recomienda un control cuidadoso de la respuesta ovárica.
En procedimientos de ART, el riesgo de embarazo múltiple depende principalmente del número de embriones transferidos, su calidad y la edad de la paciente.
Las pacientes deben informarse sobre el riesgo potencial de nacimientos múltiples antes de iniciar el tratamiento.
Interrupción del embarazo
En mujeres sometidas a estimulación del crecimiento folicular para inducir la ovulación o en ART, la frecuencia de interrupciones del embarazo por aborto espontáneo es mayor que tras la fecundación natural.
Embarazo ectópico
Las mujeres con antecedentes de enfermedad tubárica tienen riesgo de embarazo ectópico, independientemente de que este ocurra por fecundación espontánea o tras tratamiento de infertilidad. Se ha notificado que la prevalencia de embarazo ectópico tras ART es mayor que en la población general.
Neoplasias del sistema reproductivo
Se han notificado casos de tumores tanto benignos como malignos de los ovarios y otros órganos reproductivos en mujeres que han recibido múltiples medicamentos para el tratamiento de la infertilidad. Aún no se ha determinado si el tratamiento con gonadotropinas incrementa el riesgo basal de desarrollar estos tumores en mujeres infértiles.
Anomalías congénitas
La prevalencia de anomalías congénitas tras ART puede ser ligeramente mayor que tras la fecundación espontánea. Se considera que esto es consecuencia de diferencias en las características de los padres (por ejemplo, edad materna, calidad del esperma) y del embarazo múltiple.
Fenómenos tromboembólicos
En mujeres con antecedentes recientes o actuales de enfermedades tromboembólicas, o en aquellas con factores de riesgo establecidos para fenómenos tromboembólicos (como antecedentes personales o familiares), el tratamiento con gonadotropinas puede aumentar aún más el riesgo de empeoramiento o aparición de estos eventos. En tales pacientes, debe evaluarse el beneficio del uso de gonadotropinas frente al riesgo existente de estas complicaciones. Sin embargo, debe señalarse que tanto el embarazo como el OHSS aumentan el riesgo de complicaciones tromboembólicas.
Tratamiento de hombres
El aumento de los niveles endógenos de FSH en los pacientes indica una insuficiencia testicular primaria. Estos pacientes son insensibles al tratamiento con GONAL-f*/hCG. No debe administrarse GONAL-f* cuando no se puede obtener una respuesta terapéutica efectiva.
Para evaluar la respuesta al tratamiento, se recomienda realizar un análisis de semen entre 4 y 6 meses después del inicio del tratamiento.
Contenido de sodio
GONAL-f* contiene menos de 1 mmol de sodio (23 mg) por dosis, por lo tanto, es prácticamente libre de sodio.
Uso durante el embarazo o la lactancia.
Embarazo
No existen indicaciones para el uso de GONAL-f* durante el embarazo. Los datos obtenidos de un número reducido de casos de uso del medicamento durante el embarazo (menos de 300 casos) indican ausencia de malformaciones congénitas o toxicidad feto-neonatal por folitropina alfa, aunque los datos clínicos son insuficientes para descartar un efecto teratogénico de GONAL-f*.
Lactancia
GONAL-f* no está indicado para su uso durante la lactancia.
Fertilidad
GONAL-f* está indicado para el tratamiento de la infertilidad (ver sección «Indicaciones»).
Capacidad para conducir vehículos y manejar maquinaria.
GONAL-f* no afecta o afecta mínimamente la capacidad de los pacientes para conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
La administración del medicamento GONAL-f® debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la infertilidad.
Las dosis diarias, la pauta de administración y los procedimientos de monitorización del tratamiento con GONAL-f® deben individualizarse para cada paciente con el fin de optimizar el desarrollo folicular y minimizar el riesgo de hiperestimulación ovárica no deseada. Se recomienda seguir las dosis iniciales propuestas a continuación.
Se ha demostrado la bioequivalencia entre las formas farmacéuticas de dosis única y múltiples de GONAL-f®.
Mujeres con anovulación, incluyendo el síndrome de ovario poliquístico
GONAL-f® se administra en forma de un curso de inyecciones diarias. En pacientes con menstruación, el tratamiento debe iniciarse durante los primeros 7 días del ciclo menstrual.
En estudios clínicos registrados, el régimen de tratamiento habitualmente empleado comenzaba con la administración diaria de 75-150 UI de FSH. Si fuera necesario, la dosis del medicamento podía aumentarse en 37,5 UI (preferiblemente) o en 75 UI, con intervalos de 7 días o (preferiblemente) de 14 días, con el fin de obtener una respuesta adecuada pero no excesiva.
En la práctica clínica, la dosis inicial se selecciona generalmente de forma individual según los parámetros clínicos de la paciente, tales como marcadores de reserva ovárica, edad, índice de masa corporal y, si procede, la respuesta previa de los ovarios a la estimulación.
Dosis inicial
La dosis inicial puede ajustarse progresivamente de la siguiente manera: (a) menos de 75 UI por día, si se espera una respuesta ovárica excesiva basándose en el perfil clínico de la paciente (edad, índice de masa corporal, reserva ovárica), o (b) más de 75 UI hasta un máximo de 150 UI por día, si se espera una respuesta ovárica débil.
La respuesta de la paciente al tratamiento debe monitorizarse cuidadosamente mediante ecografía para medir el tamaño y número de folículos y/o mediante la medición de los niveles séricos de estrógenos.
Ajuste de la dosis
Si la paciente no responde adecuadamente al tratamiento durante 4 semanas (presentando una respuesta ovárica débil o excesiva), se debe evaluar la conveniencia de continuar con ese ciclo de tratamiento y actuar según los protocolos estándar. En caso de respuesta débil, la dosis diaria no debe exceder las 225 UI de FSH.
Si el médico observa una respuesta ovárica excesiva, el tratamiento debe interrumpirse y debe cancelarse la administración de LH (véase la sección «Precauciones de uso»). En el siguiente ciclo de tratamiento, se debe comenzar con una dosis inferior a la empleada en el ciclo anterior.
Maduración final de los folículos
Al alcanzar una respuesta ovárica óptima, se administra una sola dosis de 250 mcg de gonadotropina coriónica humana recombinante (r-hCG) o de 5000-10000 UI de hCG, 24-48 horas después de la última inyección de GONAL-f®. Se recomienda que la paciente tenga relaciones sexuales el día de la administración de hCG y el día siguiente. Alternativamente, puede realizarse una inseminación intrauterina.
Estimulación del desarrollo de múltiples folículos en mujeres sometidas a superovulación en el marco de técnicas de reproducción asistida o fecundación in vitro
En estudios clínicos registrados, el régimen habitual para la superovulación consistía en la administración diaria de 150-225 UI de GONAL-f®, comenzando en el día 2 o 3 del ciclo.
En la práctica clínica, la dosis inicial generalmente se individualiza según parámetros clínicos de la paciente, como marcadores de reserva ovárica, edad, índice de masa corporal y, si procede, la respuesta previa de los ovarios a la estimulación.
Dosis inicial
Si se espera una respuesta ovárica débil, la dosis inicial puede ajustarse progresivamente, pero no debe exceder las 450 UI. Por el contrario, si se espera una respuesta ovárica excesiva, la dosis inicial puede reducirse por debajo de 150 UI.
La respuesta de la paciente al tratamiento debe monitorizarse cuidadosamente mediante ecografía para medir el tamaño y número de folículos y/o mediante la medición de los niveles séricos de estrógenos, hasta alcanzar un desarrollo folicular adecuado.
GONAL-f® puede administrarse de forma aislada o en combinación con un agonista o antagonista de la hormona liberadora de gonadotropinas (GnRH) para prevenir la luteinización prematura.
Ajuste de la dosis
Si la paciente no responde adecuadamente al tratamiento (presentando una respuesta ovárica débil o excesiva), se debe evaluar la conveniencia de continuar con ese ciclo de tratamiento y actuar según los protocolos estándar. En caso de respuesta débil, la dosis diaria no debe exceder las 450 UI de FSH.
Maduración final de los folículos
Al alcanzar una respuesta ovárica óptima, para inducir la maduración final de los folículos, se administra una sola dosis de 250 mcg de r-hCG o de 5000-10000 UI de hCG, 24-48 horas después de la última inyección de GONAL-f®.
Mujeres con deficiencia severa de secreción de LH y FSH
En mujeres con deficiencia severa de secreción de LH y FSH, el objetivo del tratamiento combinado con GONAL-f® y un medicamento de hormona luteinizante (LH) es promover el desarrollo folicular seguido de la maduración final tras la administración de gonadotropina coriónica humana (hCG). GONAL-f® se administra en forma de inyecciones diarias simultáneamente con lutrópina alfa. Si la paciente presenta amenorrea y una secreción endógena baja de estrógenos, el tratamiento puede iniciarse en cualquier momento.
El régimen recomendado comienza con la administración diaria de 75 UI de lutrópina alfa junto con 75-150 UI de FSH. El tratamiento debe adaptarse a la respuesta individual de la paciente, evaluada mediante ecografía del tamaño folicular y niveles séricos de estrógenos.
Si se considera necesario aumentar la dosis de FSH, se recomienda hacerlo con intervalos de 7-14 días, incrementando 37,5-75 UI. Se permite prolongar la duración de la estimulación hasta 5 semanas dentro de un mismo ciclo de tratamiento.
Al alcanzar una respuesta óptima, se administra una sola dosis de 250 mcg de r-hCG o 5000-10000 UI de hCG, 24-48 horas después de la última inyección de GONAL-f® y lutrópina alfa. Se recomienda que la paciente tenga relaciones sexuales el día de la administración de hCG y el día siguiente. Alternativamente, puede realizarse una inseminación intrauterina u otra técnica de reproducción asistida según la decisión individual del médico.
Durante el tratamiento, debe considerarse la necesidad de soporte de la fase lútea, ya que la deficiencia de sustancias con actividad luteotrópica (LH/hCG) tras la ovulación puede provocar una insuficiencia prematura del cuerpo lúteo.
Si se observa una respuesta excesiva, el tratamiento debe interrumpirse y debe cancelarse la administración de hCG. En el siguiente ciclo de tratamiento, se debe comenzar con una dosis de FSH inferior a la empleada en el ciclo anterior (véase la sección «Precauciones de uso»).
Hombres con hipogonadismo hipogonadotrópico
GONAL-f® se administra a una dosis de 150 UI tres veces por semana, simultáneamente con hCG, durante al menos 4 meses. Si tras este período no se observa respuesta, el tratamiento combinado puede continuar. La experiencia clínica actual indica que, si es necesario, el tratamiento puede prolongarse al menos durante 18 meses para lograr la espermatogénesis.
Grupos de pacientes particulares
Pacientes de edad avanzada
No existen indicaciones apropiadas para el uso de GONAL-f® en pacientes de edad avanzada. La seguridad y eficacia del medicamento en este grupo no han sido establecidas.
Pacientes con alteración de la función renal o hepática
La seguridad, eficacia y parámetros farmacocinéticos de GONAL-f® en pacientes con alteración de la función renal o hepática no han sido establecidos.
Si el paciente se administra GONAL-f® por sí mismo, debe leer y seguir las instrucciones siguientes.
Recomendaciones generales
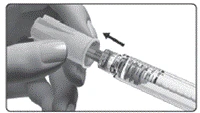
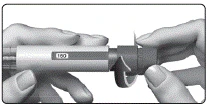
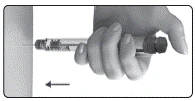
El medicamento está indicado para administración subcutánea. No debe inyectarse el medicamento si la solución es opaca o contiene partículas.
Solo los pacientes adecuadamente entrenados pueden autoinyectarse, y deben tener acceso a consultas médicas si fuera necesario.
Se recomienda realizar las inyecciones diarias a la misma hora del día, cambiando cada vez el sitio de inyección. Debe asegurarse de que los pacientes dispongan siempre del número suficiente de dispositivos de administración previstos en el plan de tratamiento.
Debe preparar el dispositivo precargado con GONAL-f® para la administración e inyectar la dosis prescrita, tal como se describe a continuación. El número que aparece en la ventana de control de dosis indica la cantidad de unidades internacionales (UI) de fólitropina alfa.
Inmediatamente después de la inyección, debe retirar la aguja del dispositivo de administración.
No se deben reutilizar las agujas. El dispositivo de administración y/o las agujas no deben compartirse con otras personas.
No debe utilizarse el dispositivo de administración si ha caído, está agrietado o dañado, ya que su uso podría provocar lesiones.
Llevar un registro diario de dosificación
Utilice un registro diario para anotar las dosis administradas cada día. La administración de una dosis incorrecta puede perjudicar su tratamiento.
- Anote el día del tratamiento (1), la fecha (2) y la hora (3) de cada inyección, así como el dispositivo de administración utilizado (4).
- Anote la dosis prescrita en la columna (5).
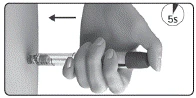
- Antes de la inyección, verifique que ha seleccionado la dosis correcta (6).
- Tras la inyección, compruebe el número en la ventana de control de dosis.
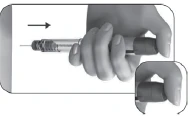
- Confirme que ha administrado la inyección completa (7), o anote el número mostrado en la ventana de control de dosis si no es «0» (8).
- Si fuera necesario, administre una segunda inyección con otro dispositivo, estableciendo la dosis indicada en la columna «Dosis a establecer para la segunda inyección» (8).
- Anote esta dosis pendiente en la columna «Dosis a administrar» de la siguiente fila (columna 6).
Ejemplo de registro diario de dosificación
| Día de tratamiento |
Fecha |
Hora |
Dosificación de la pluma |
Dosis prescrita |
Visor de control de dosis |
||||||||||||||||||||||||||||
| Dosis para la inyección |
Dosis que debe ajustarse para la segunda inyección |
||||||||||||||||||||||||||||||||
| 1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
||||||||||||||||||||||||||
| № 1 |
10 de junio |
7:00 |
300 UI |
125 |
125 |
√ Si es 0, la inyección está completada |
□ Si no es 0, se requiere una segunda inyección; Administrar _____ con una nueva pluma |
||||||||||||||||||||||||||
| № 2 |
11 de junio |
7:00 |
300 UI |
125 |
125 |
√ Si es 0, la inyección está completada |
□ Si no es 0, se requiere una segunda inyección; Administrar _____ con una nueva pluma |
||||||||||||||||||||||||||
| № 3 |
12 de junio |
7:00 |
300 UI |
125 |
125 |
□ Si es 0, la inyección está completada |
√ Si no es 0, se requiere una segunda inyección; Administrar 75 con una nueva pluma |
||||||||||||||||||||||||||
| № 3 |
12 de junio |
7:00 |
300 UI |
- |
75
No utilice el horno microondas ni otros dispositivos de calentamiento para calentar la solución.
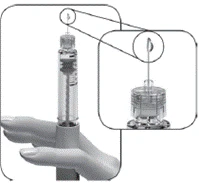
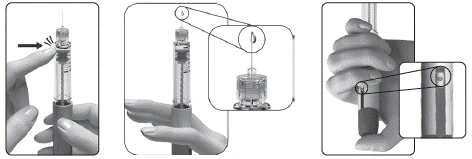
Si durante el primer uso de una nueva pluma no ve pequeñas gotas de líquido en la punta de la aguja:
Nota: La pluma precargada contiene 300 UI, 450 UI o 900 UI de folitropina alfa. La dosis máxima por administración que puede seleccionarse es de 300 UI (para la pluma de 300 UI) o 450 UI (para las plumas de 450 UI y 900 UI). La dosis mínima por administración que puede seleccionarse es de 12,5 UI.
No conserve el bolígrafo con la aguja acoplada, ya que podría provocar una infección. Niños. No existen indicaciones adecuadas para el uso de GONAL-f® en pacientes pediátricos. Sobredosificación. Los efectos de la sobredosificación con GONAL-f® son desconocidos, aunque existe la posibilidad de desarrollar el síndrome de hiperestimulación ovárica, descrito en la sección «Precauciones de uso». Reacciones adversas.Descripción general del perfil de seguridad Las reacciones adversas más frecuentemente notificadas durante el tratamiento con este medicamento son cefalea, quistes ováricos y reacciones locales en el lugar de inyección (por ejemplo, dolor, eritema, hematoma, edema y/o irritación en el lugar de inyección). Con frecuencia se han notificado casos de síndrome de hiperestimulación ovárica (SHO) de grado leve o moderado, que debe considerarse un riesgo inherente del procedimiento de estimulación. Las formas graves de SHO son infrecuentes. Muy raramente se han descrito casos de tromboembolismo (ver sección «Precauciones de uso»). Lista de reacciones adversas Para la clasificación de la frecuencia de las reacciones adversas se utiliza la siguiente terminología: muy frecuentes (≥ 1/10); frecuentes (de ≥ 1/100 a < 1/10); poco frecuentes (de ≥ 1/1000 a < 1/100); raras (de ≥ 1/10000 a < 1/1000); muy raras (< 1/10000). Tratamiento en mujeres Trastornos del sistema inmunitario Muy raras: reacciones de hipersensibilidad de intensidad leve a grave, incluyendo reacciones anafilácticas y shock. Trastornos del sistema nervioso Muy frecuentes: cefalea. Trastornos vasculares Muy raras: tromboembolismo (relacionado y no relacionado con el SHO). Trastornos del sistema respiratorio Muy raras: empeoramiento o exacerbación del asma. Trastornos gastrointestinales Frecuentes: dolor abdominal, sensación de distensión y molestias abdominales, náuseas, vómitos, diarrea. Trastornos del sistema reproductor y de las glándulas mamarias Muy frecuentes: quistes ováricos; frecuentes: SHO de grado leve a moderado (incluyendo síntomas asociados); poco frecuentes: SHO grave (incluyendo síntomas asociados) (ver sección «Precauciones de uso»); raras: complicaciones del SHO grave. Trastornos generales y en el lugar de administración Muy frecuentes: reacciones en el lugar de inyección (por ejemplo, dolor, eritema, hematoma, edema y/o irritación en el lugar de inyección). Tratamiento en hombres Trastornos del sistema inmunitario Muy raras: reacciones de hipersensibilidad de intensidad leve a grave, incluyendo reacciones anafilácticas y shock. Trastornos del sistema respiratorio Muy raras: empeoramiento o exacerbación del asma. Trastornos de la piel y del tejido subcutáneo Frecuentes: acné. Trastornos del sistema reproductor y de las glándulas mamarias Frecuentes: ginecomastia, varicocele. Trastornos generales y en el lugar de administración Muy frecuentes: reacciones en el lugar de inyección (por ejemplo, dolor, eritema, hematoma, edema y/o irritación en el lugar de inyección). Otras Frecuentes: aumento de peso. Período de validez. 2 años. No utilizar después de la fecha de caducidad indicada en el envase. El paciente debe anotar la fecha del primer uso del bolígrafo precargado con GONAL-f®. Antes de abrirlo y dentro del período de validez, el medicamento puede conservarse sin refrigeración a una temperatura máxima de 25 °C durante un período único de hasta 3 meses. El medicamento debe desecharse si no se ha utilizado dentro de estos 3 meses. Después de abrirlo, el medicamento debe conservarse a una temperatura de 2 - 25 °C durante un máximo de 28 días. Condiciones de conservación. Conservar a una temperatura de 2 - 8 ºC (en el refrigerador). No congelar. Para protegerlo de la luz, el bolígrafo de administración debe mantenerse con la tapa colocada. Mantener fuera del alcance de los niños. Envase.
Categoría de dispensación. Bajo receta médica. Fabricante. Merk Serono S.p.A./Merck Serono S.p.A. Domicilio del fabricante y dirección de su lugar de actividad. Via delle Magnolie 15 (localidad Zona Industriale), 70026 Modugno (Bari), Italia / Via delle Magnolie 15 (loc. frazione Zona Industriale), 70026 Modugno (Bari), Italy. | ||||||||||||||||||||||||||||