Favipiravir-Mikrokhim
Ucrania
Contenido
- INSTRUCCIÓN PARA USO MÉDICO DEL MEDICAMENTO FAVIPIRAVIR-MICROKHIM (FAVIPIRAVIR-MICROKHIM)
- Composición:
- Propiedades farmacológicas.
- Características clínicas.
- Características de uso.
- Vía de administración y dosis.
- Reacciones adversas.
- Composición:
- Propiedades farmacológicas.
- Características clínicas.
- Características de uso.
- Vía de administración y dosis.
- Reacciones adversas.
¡ATENCIÓN! ¡ADVERTENCIA! El medicamento tiene un efecto perjudicial cuando se utiliza durante el embarazo y puede causar la muerte del embrión y/o efectos teratogénicos sobre el feto. Está contraindicado durante el embarazo y la lactancia. Durante la administración del medicamento y durante 7 días después de finalizar el tratamiento, las parejas sexuales deben utilizar métodos anticonceptivos altamente eficaces; los hombres deben usar condones. |
INSTRUCCIÓN PARA USO MÉDICO DEL MEDICAMENTO FAVIPIRAVIR-MICROKHIM (FAVIPIRAVIR-MICROKHIM)
Composición:
Principio activo: favipiravir;
1 tableta contiene 200 mg de favipiravir;
Excipientes: hidroxipropilcelulosa de bajo grado de sustitución, povidona, dióxido de silicio coloidal anhidro, crospovidona, estearilfumarato sódico, hipromelosa, polietilenglicol 6000 (macrogol 6000), talco, dióxido de titanio (E 171), óxido de hierro amarillo (E 172).
Forma farmacéutica. Tabletas recubiertas con película.
Propiedades físico-químicas principales: tabletas redondas, biconvexas, de color amarillo, recubiertas con película.
Grupo farmacoterapéutico.
Medicamentos antivirales para uso sistémico. Antivirales de acción directa. Otros medicamentos antivirales. Favipiravir. Código ATC J05AX27.
Propiedades farmacológicas.
Farmacodinamia.
Actividad antiviral in vitro
El favipiravir demostró actividad antiviral frente a cepas de laboratorio del virus de la gripe tipo A y tipo B, con una concentración efectiva semimáxima (CE50) de 0,014–0,55 μg/ml.
La CE50 frente a virus de la gripe estacionales tipo A y tipo B, incluyendo cepas resistentes a adamantanos (amantadina y rimantadina), oseltamivir o zanamivir, fue de 0,03–0,94 y 0,09–0,83 μg/ml, respectivamente.
La CE50 frente a virus de la gripe tipo A y tipo B resistentes a adamantanos, oseltamivir y zanamivir fue de 0,09–0,47 μg/ml, sin observarse resistencia cruzada.
La CE50 frente a virus de la gripe tipo A (incluyendo cepas resistentes a adamantanos, oseltamivir o zanamivir), tales como gripe A porcina y gripe A aviar, incluyendo cepas de alta patogenicidad (incluyendo H5N1 y H7N9), fue de 0,06–3,53 μg/ml.
Mecanismo de acción
El favipiravir se metaboliza en las células hasta la forma de ribosil trifosfato (RTP) del favipiravir y de forma selectiva inhibe la ARN polimerasa implicada en la replicación del virus de la gripe. El RTP del favipiravir (1000 μmol/l) no mostró actividad inhibitoria sobre el ADN α humano, pero mostró actividad inhibitoria en el rango del 9,1–13,5 % sobre el ADN β y en el rango del 11,7–41,2 % sobre el ADN γ humano. La concentración inhibitoria (CI50) del RTP del favipiravir para la ARN polimerasa II humana fue de 905 μmol/l.
Resistencia
No se observaron cambios en la sensibilidad de los virus de la gripe tipo A al favipiravir, ni se detectaron virus resistentes. En los estudios clínicos realizados, incluyendo el estudio global de Fase III, no se observó aparición de virus de la gripe resistentes al favipiravir.
Farmacocinética.
Absorción
Concentración en sangre
En la tabla siguiente se presentan los parámetros farmacocinéticos del favipiravir administrado por vía oral a 8 adultos sanos en una dosis de 1600 mg dos veces al día el primer día, seguido de 600 mg dos veces al día durante 4 días (1600 mg/600 mg dos veces al día) y posterior administración única de una dosis de 600 mg una vez al día.
Parámetros farmacocinéticos del favipiravir
| Dosificación |
Día |
Cmax (μg/ml) |
AUC (μg·h/ml) |
Tmax (h) |
T1/2 (h) |
| 1600 mg/600 mg dos veces al día |
Días 1-5 |
64,56 (17,2) |
446,09 (28,1) |
1,5 (0,75; 4) |
4,8 ± 1,1 |
| 600 mg una vez al día |
día 6 |
64,69 (24,1) |
553,98 (31,2) |
1,5 (0,75; 2) |
5,6 ± 2,3 |
Cambios en la concentración media de favipiravir en plasma (media ± desviación estándar)
 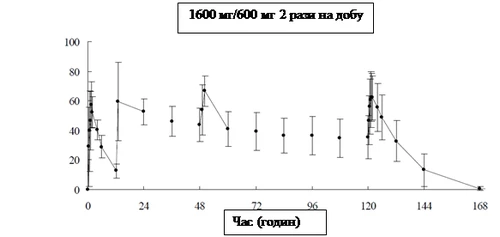 |
Después de la administración oral repetida de favipiravir durante 7 días en voluntarios sanos con actividad mínima de aldehído oxidasa (AO), el valor calculado del AUC del fármaco sin cambios fue de 1452,73 μg·h/mL en el día 1 y de 1324,09 μg·h/mL en el día 7.
Distribución
Cuando 20 hombres adultos sanos recibieron favipiravir por vía oral a una dosis de 1200 mg dos veces al día durante el primer día y luego 800 mg dos veces al día durante 4 días, el valor medio geométrico de la concentración del fármaco en el semen fue de 18,341 μg/mL en el día 3 de tratamiento y de 0,053 μg/mL en el día 2 después de la interrupción del tratamiento. A los 7 días después de la interrupción del tratamiento, los niveles del fármaco en el semen estuvieron por debajo del límite cuantificable (0,02 μg/mL) en todos los participantes del estudio. La relación media de la concentración del fármaco en el semen respecto a su concentración en plasma fue de 0,53 en el día 3 de tratamiento y de 0,45 en el día 2 tras la interrupción del tratamiento. El coeficiente de unión a proteínas en suero sanguíneo fue de 53,4–54,4 % (in vitro, mediante ultracentrifugación con filtro) a concentraciones plasmáticas del fármaco entre 0,3 y 30 μg/mL.
Metabolismo
El favipiravir no es metabolizado por el citocromo P450 (CYP); principalmente es metabolizado por la AO y parcialmente por la xantina oxidasa (XO) a su forma hidroxilada. En estudios con microsomas hepáticos humanos, la formación del hidroxilato osciló entre 3,98 y 47,6 μmol/mg proteína/min, con una variación máxima de la actividad de AO de 12 veces. Además del metabolito hidroxilado, se observó también en plasma y orina el conjugado de glucurónido.
Excreción
El favipiravir se elimina principalmente por riñón en forma del metabolito activo, la forma hidroxilada, aunque también se observa una pequeña cantidad del fármaco sin cambios. En un estudio de 7 días con administración oral múltiple de favipiravir, el coeficiente total de excreción del fármaco sin cambios y de la forma hidroxilada fue del 0,8 % y del 53,1 %, respectivamente, durante las 48 horas posteriores a la última dosis.
Características clínicas.
Indicaciones.
Para el tratamiento de infecciones pandémicas nuevas o recurrentes causadas por el virus de la influenza en las que el tratamiento con otros agentes antivirales ha sido ineficaz o insuficientemente eficaz.
Contraindicaciones.
- Mujeres embarazadas o con sospecha de embarazo (se observó muerte embrionaria temprana y teratogenicidad en estudios en animales (ver sección «Uso durante el embarazo o la lactancia»));
- pacientes con hipersensibilidad a cualquiera de los componentes del medicamento.
Interacción con otros medicamentos y otros tipos de interacciones.
El favipiravir no es metabolizado por el citocromo P450 (CYP). Principalmente es metabolizado por AO y parcialmente por KO. El medicamento inhibe AO y CYP2C8, pero no induce CYP (ver sección «Farmacocinética»).
El favipiravir debe administrarse con precaución cuando se utiliza simultáneamente con los siguientes medicamentos.
| Medicamentos |
Signos, síntomas y tratamiento |
Mecanismo de acción y factores de riesgo |
| Pirazinamida |
Aumento de la concentración de ácido úrico en sangre. Al administrar 1,5 g de pirazinamida una vez al día y 1200 mg/400 mg de favipiravir dos veces al día, el nivel de ácido úrico en sangre fue de 11,6 mg/dl con pirazinamida sola y de 13,9 mg/dl en combinación con favipiravir. |
Aumento de la reabsorción del ácido úrico en los túbulos renales debido a un efecto aditivo. |
| Repaglinida |
Puede producirse un aumento de los niveles sanguíneos de repaglinida, con el consiguiente desarrollo de reacciones adversas a este medicamento. |
La inhibición de CYP2C8 puede provocar un aumento de la concentración sanguínea de repaglinida. |
| Teofilina |
Puede producirse un aumento de la concentración sanguínea de favipiravir y posiblemente el desarrollo de reacciones adversas a favipiravir. |
La interacción con CO puede provocar un aumento de la concentración sanguínea de favipiravir. |
| Famciclovir, sulindaco |
La eficacia de famciclovir y sulindaco puede verse reducida. |
La inhibición por favipiravir de AO puede provocar una disminución de la concentración de las formas activas de estos medicamentos en sangre. |
In vitro. El favipiravir inhibe de forma irreversible la AO de manera dependiente de la dosis y del tiempo, e inhibe el CYP2C8 de manera dependiente de la dosis. No se observó actividad inhibitoria sobre el CO, y se observó una débil actividad inhibitoria sobre el CYP1A2, 2C9, 2C19, 2D6, 2E1 y 3A4. El metabolito hidroxilado muestra una débil actividad inhibitoria sobre el CYP1A2, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4.
No se observa efecto inductor del favipiravir sobre el CYP.
Estudios clínicos de interacciones medicamentosas
Efecto de los medicamentos concomitantes sobre la farmacocinética del favipiravir
| Medicamento concomitante y dosis |
Dosificación de favipiravir |
n |
Momento de la toma de muestras |
Relación de parámetros para favipiravir (intervalo de confianza del 90 % (IC)) (administración concomitante/administración aislada) |
|
| Cmax |
AUC |
||||
| Teofilina 200 mg dos veces al día del día 1 al 9, 200 mg una vez al día el día 10 |
600 mg dos veces al día el día 6, 600 mg una vez al día del día 7 al 10 |
10 |
Día 6 |
1,33 [1,19; 1,48] |
1,27 [1,15; 1,40] |
| Día 7 |
1,03 [0,92; 1,15] |
1,17 [1,04; 1,31] |
|||
| Oseltamivir 75 mg dos veces al día del día 1 al 5, 75 mg una vez al día el día 6 |
600 mg dos veces al día el día 5, 600 mg una vez al día el día 6 |
10 |
Día 6 |
0,98 [0,87; 1,10] |
1,01 [0,91; 1,11] |
| Raloxifeno 60 mg 1 vez al día del día 1 al 3 |
1200 mg 2 veces al día el día 1, 800 mg 2 veces al día el día 2, 800 mg 1 vez al día el día 3 |
17 |
Día 1 |
1,00 [0,90; 1,10] |
1,03 [0,95; 1,12] |
| Día 3 |
0,90 [0,81; 0,99] |
0,85 [0,79; 0,93] |
|||
| Hidralazina 5 mg 1 vez al día el día 1 y el día 5 |
1200 mg/400 mg el día 1, 400 mg 2 veces al día del día 2 al 4, 400 mg 1 vez al día el día 5 |
14 |
Día 1 |
0,99 [0,92; 1,06] |
0,99 [0,92; 1,07] |
| Día 5 |
0,96 [0,89; 1,04] |
1,04 [0,96; 1,12] |
Influencia del favipiravir en la farmacocinética de los medicamentos concomitantes
| Medicamento concomitante y dosis |
Dosificación de favipiravir |
n |
Momento de la toma |
Relación de parámetros para medicamentos concomitantes (IC del 90 %) (aplicación conjunta/aplicación separada) |
|
| Cmax |
AUC |
||||
| Teofilina 200 mg dos veces al día del día 1 al 9, 200 mg una vez al día el día 10 |
600 mg dos veces al día el día 6, 600 mg una vez al día del día 7 al 10 |
10 |
Día 7 |
0,93 [0,85; 1,01] |
0,92 [0,87; 0,97] |
| Día 10 |
0,99 [0,94; 1,04] |
0,97 [0,91; 1,03] |
|||
| Oseltamivir 75 mg dos veces al día del día 1 al 5, 75 mg una vez al día el día 6 |
600 mg dos veces al día el día 5, 600 mg una vez al día el día 6 |
10 |
Día 6 |
1,10 [1,06; 1,15] |
1,14 [1,10; 1,18] |
| Paracetamol 650 mg una vez al día el día 1 y el día 5 |
1200 mg dos veces al día el día 1, 800 mg dos veces al día del día 2 al 4, 800 mg una vez al día el día 5 |
28 |
Día 1 |
1,03 [0,93; 1,14] |
1,16 [1,08; 1,25] |
| Día 5 |
1,08 [0,96; 1,22] |
1,14 [1,04; 1,26] |
|||
| Noretindrona/etinilestradiol 1 mg/0,035 mg una vez al día del día 1 al 4 |
1200 mg dos veces al día el día 1, 800 mg dos veces al día del día 2 al 4, 800 mg una vez al día el día 5 |
25 |
Día 12 |
1,23 [1,16; 1,30] |
1,47 [1,42; 1,52] |
| Día 12 |
1,48 [1,42; 1,54] |
1,43 [1,39; 1,47] |
|||
| Repaglinida 0,5 mg una vez al día el día 13 |
1200 mg dos veces al día el día 1, 800 mg dos veces al día del día 2 al 4, 800 mg una vez al día el día 5 |
17 |
Día 13 |
1,28 [1,16; 1,41] |
1,52 [1,37; 1,68] |
| Hidralazina 5 mg una vez al día el día 1 y el día 5 |
1200 mg/400 mg el día 1, 400 mg dos veces al día del día 2 al 4, 400 mg una vez al día el día 5 |
14 |
Día 1 |
0,73 [0,67; 0,81] |
0,87 [0,78; 0,97] |
| Día 5 |
0,79 [0,71; 0,88] |
0,91 [0,82; 1,01] |
|||
Características de uso.
La administración del medicamento FAVIPIRAVIR-MIKROKIM para el tratamiento del virus de la gripe solo es posible en ausencia de efecto de otra terapia y cuando el beneficio esperado para el paciente supere el riesgo.
Advertencia
Dado que en estudios en animales se han observado casos de muerte embrionaria en fases tempranas del desarrollo y efectos teratogénicos, no se debe administrar el medicamento a mujeres con embarazo confirmado o sospechado (véase la sección «Contraindicaciones» y «Uso durante el embarazo o la lactancia»).
Al administrar el medicamento a mujeres en edad fértil, antes del inicio del tratamiento se debe confirmar un resultado negativo en la prueba de embarazo. Se debe informar detalladamente a las mujeres sobre los riesgos asociados con el uso de este medicamento y recomendar firmemente el uso de métodos anticonceptivos altamente eficaces tanto por parte de la mujer como de su pareja durante el tratamiento con este fármaco y durante 7 días posteriores a la finalización del mismo (véase la sección «Uso durante el embarazo o la lactancia»). Si durante el tratamiento surge una sospecha de embarazo, se debe recomendar a la mujer que interrumpa inmediatamente la toma del medicamento y consulte con su médico.
El favipiravir penetra en el semen. Al administrar el medicamento a pacientes del sexo masculino, se debe informar detalladamente sobre los riesgos asociados con el tratamiento y recomendar firmemente el uso de métodos anticonceptivos eficaces durante las relaciones sexuales durante el tratamiento y durante 7 días posteriores a su finalización (los hombres deben usar condones). Además, se debe informar a los pacientes del sexo masculino de que no deben tener relaciones sexuales con mujeres embarazadas (véanse las secciones «Uso durante el embarazo o la lactancia» y «Farmacocinética»).
Antes del inicio del tratamiento, se debe proporcionar información detallada sobre la eficacia del medicamento y sus riesgos (incluido el riesgo de efectos sobre el feto).
Antes de iniciar el tratamiento con el medicamento, se debe evaluar cuidadosamente la conveniencia de su uso.
Precauciones
FAVIPIRAVIR-MIKROKIM es un medicamento cuya administración solo debe considerarse en caso de brotes de nuevas infecciones o reaparición de infecciones causadas por el virus de la gripe, cuando otros medicamentos antivirales sean ineficaces o insuficientemente eficaces. Durante el tratamiento con este medicamento, se debe consultar la información más reciente y administrar el fármaco únicamente a pacientes adecuados.
El favipiravir no es eficaz contra infecciones bacterianas.
El favipiravir no debe administrarse a niños (véase la sección «Niños»).
Independientemente de la vía de administración o del tipo de medicamentos antivirales contra la gripe, se han registrado casos de comportamiento anómalo en pacientes con infección por el virus de la gripe (véase la sección «Reacciones adversas»). Para prevenir accidentes, como caídas debido a comportamiento inusual, como medida preventiva, se debe instruir a los pacientes o a sus cuidadores sobre lo siguiente:
- puede presentarse comportamiento anómalo,
- cuando los pacientes sean tratados en casa, los cuidadores u otras personas deben tomar medidas preventivas contra accidentes, como caídas, durante al menos 2 días tras el inicio de la fiebre.
Formas graves de comportamiento anómalo que han provocado accidentes por caídas se han observado con mayor frecuencia en niños en edad escolar y adolescentes. Se sabe que tales síntomas aparecen principalmente dentro de los 2 días posteriores al inicio de la fiebre.
Se debe observar cuidadosamente a los pacientes y, si se observan alteraciones, se debe interrumpir el tratamiento y tomar las medidas adecuadas.
La infección causada por el virus de la gripe puede complicarse con infecciones bacterianas o presentarse con síntomas que pueden confundirse fácilmente con los síntomas gripales. En caso de infección bacteriana o sospecha de infección bacteriana, se deben tomar medidas adecuadas, como la administración de antibacterianos.
Uso durante el embarazo o la lactancia.
Embarazo
Está contraindicado el uso del medicamento FAVIPIRAVIR-MIKROKIM en mujeres con embarazo conocido o sospechado. En estudios en animales con niveles de exposición similares o inferiores a la exposición clínica, se observó muerte embrionaria en fases tempranas (en ratas) y efectos teratogénicos (en monos, ratones, ratas y conejos).
Lactancia
Al administrar el medicamento FAVIPIRAVIR-MIKROKIM a mujeres que amamantan, se debe suspender la lactancia. Durante los estudios se ha demostrado que el metabolito principal del favipiravir, la forma hidroxilada, penetra en la leche materna.
Dado que en estudios en animales se han observado casos de muerte embrionaria en fases tempranas del desarrollo y efectos teratogénicos, no se debe administrar favipiravir a mujeres con embarazo confirmado o sospechado (véase la sección «Contraindicaciones»).
Al administrar favipiravir a mujeres en edad fértil, antes del inicio del tratamiento se debe confirmar un resultado negativo en la prueba de embarazo. Se debe informar detalladamente a las mujeres sobre los riesgos asociados con el uso de este medicamento y recomendar firmemente el uso de métodos anticonceptivos altamente eficaces tanto por parte de la mujer como de su pareja durante el tratamiento con este fármaco y durante 7 días posteriores a la finalización del mismo. Si durante el tratamiento surge una sospecha de embarazo, se debe recomendar a la mujer que interrumpa inmediatamente la toma del medicamento y consulte con su médico.
El favipiravir penetra en el semen. Al administrar el medicamento a pacientes del sexo masculino, se debe informar detalladamente sobre los riesgos asociados con el tratamiento y recomendar firmemente el uso de métodos anticonceptivos eficaces durante las relaciones sexuales durante el tratamiento y durante 7 días posteriores a su finalización (los hombres deben usar condones). Además, se debe informar a los pacientes del sexo masculino de que no deben tener relaciones sexuales con mujeres embarazadas (véase la sección «Farmacocinética»).
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
No se dispone de información sobre el efecto del favipiravir sobre la velocidad de reacción al conducir vehículos o manejar maquinaria.
Vía de administración y dosis.
La dosis habitual de favipiravir para adultos es de 1600 mg: se toma por vía oral dos veces al día el primer día, luego 600 mg por vía oral dos veces al día durante los siguientes 4 días.
El período total de tratamiento debe ser de 5 días.
El tratamiento debe iniciarse inmediatamente después de la aparición de los síntomas de gripe.
Grupos de pacientes especiales
Pacientes de edad avanzada (> 65 años)
Dado que en las personas de edad avanzada las funciones fisiológicas suelen estar reducidas, se debe administrar el medicamento FAVIPIRAVIR-MIKROKHIM con precaución, controlando el estado general del paciente.
Pacientes pediátricos (< 18 años)
El medicamento no está indicado para su uso en niños.
Alteraciones de la función hepática
En pacientes con alteración hepática leve y moderada (clases A y B según la clasificación de Child-Pugh, 6 personas por cada clase), tras la administración oral de favipiravir en dosis de 1200 mg dos veces al día el primer día y luego 800 mg dos veces al día durante los siguientes 4 días, en comparación con voluntarios adultos sanos, los valores de Cmax y AUC el día 5 fueron aproximadamente 1,6 veces y 1,7 veces más altos en pacientes con alteración hepática leve, y 1,4 veces y 1,8 veces más altos en pacientes con alteración hepática moderada, respectivamente.
En pacientes con alteración hepática grave (clase C según la clasificación de Child-Pugh, 4 personas), tras la administración oral de favipiravir a una dosis de 800 mg dos veces al día el primer día y luego 400 mg dos veces al día durante 2 días, en comparación con voluntarios adultos sanos, los valores de Cmax y AUC el día 3 fueron aproximadamente 2,1 y 6,3 veces más altos, respectivamente.
Situaciones que requieren precaución en la administración del medicamento
El medicamento debe administrarse con precaución a pacientes con gota activa o antecedentes de gota, así como a pacientes con hiperuricemia (en estos pacientes puede aumentar el nivel de ácido úrico en sangre, por lo que los síntomas podrían agravarse).
Niños.
El medicamento no se utiliza en la población pediátr游戏副本
Reacciones adversas.
El favipiravir no se ha administrado a la dosis autorizada. En estudios clínicos japoneses y en el estudio global de Fase III (estudios realizados con niveles de dosis inferiores a los autorizados), se observaron reacciones adversas en 100 de 501 pacientes (19,96 %) y se evaluaron desde el punto de vista de la seguridad (incluyendo valores anormales en pruebas de laboratorio).
Las principales reacciones adversas incluyeron: aumento del nivel de ácido úrico en sangre (en 24 pacientes (4,79 %)), diarrea (en 24 pacientes (4,79 %)), disminución del número de neutrófilos (en 9 pacientes (1,80 %)), elevación de AST (GOT) (en 9 pacientes (1,80 %)), aumento de ALT (GPT) (en 8 pacientes (1,60 %)).
Reacciones adversas clínicamente significativas
Aunque la relación causal es desconocida, se han notificado síntomas psiconeurológicos, tales como comportamiento anormal, tras la administración de medicamentos contra el virus de la gripe, incluyendo el favipiravir (ver sección «Instrucciones de uso»).
Reacciones adversas clínicamente significativas (observadas con medicamentos análogos)
Las siguientes reacciones adversas clínicamente significativas se han registrado con otros medicamentos utilizados contra el virus de la gripe.
Del sistema inmunitario: shock, anafilaxia.
Del sistema respiratorio, órganos del tórax y mediastino: neumonía.
Del sistema hepatobiliar: hepatitis fulminante, alteración de la función hepática, ictericia.
De la piel y del tejido subcutáneo: necrólisis epidérmica tóxica (NET), síndrome de Stevens-Johnson.
De los riñones y del sistema urinario: insuficiencia renal aguda.
Del sistema sanguíneo y sistema linfático: disminución del número de leucocitos, neutrófilos y plaquetas.
Del estado psíquico: alteración de la conciencia, delirio, alucinaciones, manía, convulsiones.
Del tubo digestivo: colitis hemorrágica.
Es necesario mantener una vigilancia constante sobre los pacientes y, en caso de aparición de reacciones adversas, interrumpir el tratamiento y adoptar las medidas adecuadas.
Otras reacciones adversas observadas en estudios clínicos japoneses y en el estudio clínico global de Fase III (estudios realizados con niveles de dosis inferiores a los autorizados)
| Clasificación de órganos y sistemas MedDRA |
Reacciones adversas |
||
| ≥ 1 % |
0,5 – < 1 % |
< 0,5 % |
|
| Hipersensibilidad |
|
Erupción cutánea |
Ecsema, prurito |
| Sistema hepatobiliar |
Aumento de AST (TGO), ALT (TGP), γ-GT |
|
Aumento de los niveles de fosfatasas alcalinas y bilirrubina en sangre |
| Tracto gastrointestinal |
Diárea (4,79 %) |
Náuseas, vómitos, dolor abdominal |
Molestias abdominales, úlcera duodenal, hematocacía, gastritis |
| Sangre y sistema linfático |
Disminución del número de neutrófilos y leucocitos |
|
Aumento del número de leucocitos y monocitos, disminución del número de reticulocitos |
| Metabolismo y trastornos nutricionales |
Aumento del ácido úrico (4,79 %) y de los triglicéridos en sangre |
Glucosuria |
Disminución del potasio en sangre |
| Sistema respiratorio, órganos torácicos y mediastino |
|
|
Asma bronquial, dolor orofaríngeo, rinitis, nasofaringitis |
| Otras |
|
|
Aumento de CK (CPK), presencia de sangre en orina, pólipo de amígdalas, pigmentación, disgeusia, equimosis, visión borrosa, dolor ocular, vértigo, extrasístoles supraventriculares |
Ante la aparición de las reacciones adversas anteriormente mencionadas, se deben tomar las medidas adecuadas de acuerdo con la sintomatología presente.
Plazo de validez.
1,5 años.
Condiciones de almacenamiento.
Conservar a una temperatura no superior a 25 °C en el envase original.
Conservar en un lugar inaccesible para los niños.
Envase.
10 comprimidos por blíster, 4 blísteres por estuche de cartón o 10 comprimidos por blíster, 10 blísteres por estuche de cartón.
O bien 40 comprimidos en frasco, 1 frasco por estuche de cartón, o 100 comprimidos en frasco, 1 frasco por estuche de cartón.
Categoría de dispensación.
Medicamento sujeto a prescripción médica.
Fabricante.
NVF S.A. «MICROKHIM».
Domicilio del fabricante y dirección del lugar de ejercicio de su actividad.
Ucrania, 93400, región de Lugansk, ciudad de Severodonetsk, calle Promyslova, 24-v.
Titular del registro.
NVF S.A. «MICROKHIM».
Domicilio del titular del registro.
Ucrania, 01013, ciudad de Kiev, calle Budindustrii, 5.
Fecha de la última revisión.
Puede informar sobre cualquier reacción adversa al medicamento llamando al teléfono +38 (050) 309-83-54 (disponible las 24 horas).
INSTRUCCIÓN
para uso médico del medicamento
FAVIPIRAVIR-MICROKHIM
(FAVIPIRAVIR-MICROKHIM)
Composición:
Principio activo: favipiravir;
1 tableta contiene 200 mg de favipiravir;
Excipientes: hidroxipropilcelulosa de bajo grado de sustitución, povidona, dióxido de silicio coloidal anhidro, crospovidona, estearilfumarato sódico, hipromelosa, polietilenglicol 6000 (macrogol 6000), talco, dióxido de titanio (E 171), óxido de hierro amarillo (E 172).
Forma farmacéutica. Tabletas recubiertas con película.
Características físicas y químicas principales: tabletas redondas, biconvexas, de color amarillo, recubiertas con película.
Grupo farmacoterapéutico.
Medicamentos antivirales para uso sistémico. Antivirales de acción directa. Otros medicamentos antivirales. Favipiravir. Código ATC J05AX27.
Propiedades farmacológicas.
Farmacodinamia.
Actividad antiviral in vitro
El favipiravir demostró actividad antiviral frente a cepas de laboratorio del virus de la gripe tipo A y tipo B, con una concentración efectiva media (CE50) de 0,014–0,55 μg/ml.
La CE50 frente a virus de la gripe estacionales tipo A y tipo B, incluyendo cepas resistentes a adamantanos (amantadina y rimantadina), oseltamivir o zanamivir, fue de 0,03–0,94 y 0,09–0,83 μg/ml, respectivamente.
La CE50 frente a virus de la gripe tipo A y tipo B resistentes a adamantanos, oseltamivir y zanamivir fue de 0,09–0,47 μg/ml, sin observarse resistencia cruzada.
La CE50 frente a virus de la gripe tipo A (incluyendo cepas resistentes a adamantanos, oseltamivir o zanamivir), tales como gripe A porcina y gripe A aviar, incluyendo cepas de alta patogenicidad (incluyendo H5N1 y H7N9), fue de 0,06–3,53 μg/ml.
Mecanismo de acción
El favipiravir se metaboliza en las células hasta la forma de ribosil trifosfato (RTP) del favipiravir y suprime selectivamente la ARN polimerasa implicada en la replicación del virus de la gripe. El RTP del favipiravir (1000 μmol/l) no mostró efecto inhibitorio sobre el ADN α humano, pero mostró un efecto inhibitorio en el rango del 9,1–13,5 % sobre el ADN β y del 11,7–41,2 % sobre el ADN γ humano. La concentración inhibitoria (CI50) del RTP del favipiravir para la ARN polimerasa II humana fue de 905 μmol/l.
Resistencia
No se observaron cambios en la sensibilidad de los virus de la gripe tipo A al favipiravir, ni se detectaron virus resistentes. En los estudios clínicos realizados, incluyendo un estudio global de Fase III, no se observó aparición de virus de la gripe resistentes al favipiravir.
Farmacocinética.
Absorción
Concentración en sangre
En la tabla siguiente se presentan los parámetros farmacocinéticos del favipiravir administrado por vía oral a 8 adultos sanos en una dosis de 1600 mg dos veces al día el primer día, seguido de 600 mg dos veces al día durante 4 días (1600 mg/600 mg dos veces al día), seguido de una dosis única de 600 mg una vez al día.
Parámetros farmacocinéticos del favipiravir
| Dosificación |
Día |
Cmax (mcg/ml) |
AUC (mcg·h/ml) |
Tmax (h) |
T1/2 (h) |
| 1600 mg/600 mg dos veces al día |
Días 1-5 |
64,56 (17,2) |
446,09 (28,1) |
1,5 (0,75; 4) |
4,8 ± 1,1 |
| 600 mg una vez al día |
día 6 |
64,69 (24,1) |
553,98 (31,2) |
1,5 (0,75; 2) |
5,6 ± 2,3 |
Cambios en la concentración media de favipiravir en plasma sanguíneo (media ± desviación estándar)
 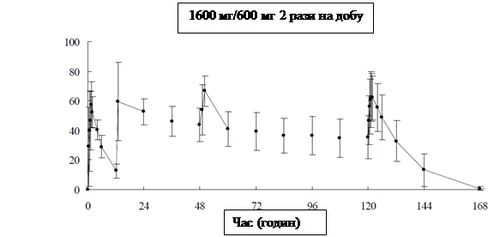 |
Después de la administración oral repetida de favipiravir durante 7 días en voluntarios sanos con actividad de aldehído oxidasa (AO) baja, el valor calculado de AUC del fármaco sin cambios fue de 1452,73 μg·h/ml en el día 1 y de 1324,09 μg·h/ml en el día 7.
Distribución
Cuando 20 hombres adultos sanos recibieron favipiravir por vía oral en dosis de 1200 mg dos veces al día durante el primer día y luego 800 mg dos veces al día durante 4 días, el valor medio geométrico de la concentración del fármaco en el semen fue de 18,341 μg/ml en el día 3 de tratamiento y de 0,053 μg/ml en el día 2 después de la interrupción del tratamiento. A los 7 días después de la interrupción del tratamiento, los niveles del fármaco en el semen estuvieron por debajo del límite de cuantificación (0,02 μg/ml) en todos los participantes del estudio. La relación media de la concentración del fármaco en el semen respecto a su concentración en plasma fue de 0,53 en el día 3 de tratamiento y de 0,45 en el día 2 tras la interrupción del tratamiento. El coeficiente de unión a proteínas en suero fue de 53,4–54,4 % (in vitro, ultrafiltración centrífuga) a concentraciones plasmáticas del fármaco entre 0,3 y 30 μg/ml.
Metabolismo
El favipiravir no es metabolizado por el citocromo P450 (CYP), siendo metabolizado principalmente por la AO y parcialmente por la xantina oxidasa (XO) a su forma hidroxilada. En estudios con microsomas hepáticos humanos, la formación del hidroxilato osciló entre 3,98 y 47,6 μmol/mg proteína/min, con una variación máxima de la actividad de AO de 12 veces. Además de la forma hidroxilada, en plasma y orina se observó también el conjugado de glucurónido como otro metabolito.
Excreción
El favipiravir se elimina principalmente por riñón en forma del metabolito activo, la forma hidroxilada, observándose también una pequeña cantidad del fármaco sin cambios. En un estudio de 7 días con administración múltiple oral de favipiravir, el coeficiente total de excreción del fármaco sin cambios y de la forma hidroxilada fue del 0,8 % y del 53,1 %, respectivamente, durante las 48 horas posteriores a la última dosis.
Características clínicas.
Indicaciones.
Para el tratamiento de infecciones pandémicas nuevas o recurrentes causadas por el virus de la gripe, en las que el tratamiento con otros agentes antivirales ha sido ineficaz o insuficientemente eficaz.
Contraindicaciones.
- Mujeres embarazadas o sospecha de embarazo (se observó muerte embrionaria temprana y teratogenicidad en estudios en animales (ver sección «Uso durante el embarazo o la lactancia»));
- pacientes con hipersensibilidad a cualquiera de los componentes del medicamento.
Interacción con otros medicamentos y otros tipos de interacciones.
El favipiravir no es metabolizado por el citocromo P450 (CYP). Principalmente es metabolizado por AO y parcialmente por KO. El medicamento inhibe AO y CYP2C8, pero no induce CYP (ver sección «Farmacocinética»).
El favipiravir debe administrarse con precaución cuando se utiliza simultáneamente con los siguientes medicamentos.
| Medicamentos |
Signos, síntomas y tratamiento |
Mecanismo de acción y factores de riesgo |
| Pirazinamida |
Aumento de la concentración de ácido úrico en sangre. Al administrar 1,5 g de pirazinamida una vez al día y 1200 mg/400 mg de favipiravir dos veces al día, el nivel de ácido úrico en sangre fue de 11,6 mg/dl con pirazinamida sola y de 13,9 mg/dl en combinación con favipiravir. |
Aumento de la reabsorción de ácido úrico en los túbulos renales debido a un efecto aditivo. |
| Repaglinida |
Puede aumentar el nivel de repaglinida en sangre, con el consiguiente desarrollo de reacciones adversas a este medicamento. |
La inhibición de CYP2C8 puede provocar un aumento de la concentración de repaglinida en sangre. |
| Teofilina |
Puede producirse un aumento de la concentración de favipiravir en sangre, con posible aparición de reacciones adversas al favipiravir. |
La interacción con CO puede provocar un aumento de la concentración de favipiravir en sangre. |
| Famciclovir, sulindac |
La eficacia del famciclovir y del sulindac puede verse reducida. |
La inhibición por favipiravir de AO puede provocar una disminución de la concentración de las formas activas de estos medicamentos en sangre. |
In vitro. El favipiravir inhibe irreversiblemente la AO de forma dependiente de la dosis y del tiempo, e inhibe CYP2C8 de forma dependiente de la dosis. No se observó actividad inhibitoria sobre CYP450 y se observó una débil actividad inhibitoria sobre CYP1A2, 2C9, 2C19, 2D6, 2E1 y 3A4. El metabolito hidroxilado muestra una débil actividad inhibitoria sobre CYP1A2, 2C8, 2C9, 2C19, 2D6, 2E1 y 3A4.
No se observa efecto inductor del favipiravir sobre CYP.
Estudios clínicos de interacciones medicamentosas
Efecto de medicamentos concomitantes sobre la farmacocinética del favipiravir
| Medicamento concomitante y dosis |
Dosificación de favipiravir |
n |
Momento de la dosificación |
Relación de parámetros para favipiravir (intervalo de confianza del 90 % (IC)) (administración conjunta/administración por separado) |
|
| Cmax |
AUC |
||||
| Teofilina 200 mg dos veces al día del día 1 al 9, 200 mg una vez al día el día 10 |
600 mg dos veces al día el día 6, 600 mg una vez al día del día 7 al 10 |
10 |
Día 6 |
1,33 [1,19; 1,48] |
1,27 [1,15; 1,40] |
| Día 7 |
1,03 [0,92; 1,15] |
1,17 [1,04; 1,31] |
|||
| Oseltamivir 75 mg dos veces al día del día 1 al 5, 75 mg una vez al día el día 6 |
600 mg dos veces al día el día 5, 600 mg una vez al día el día 6 |
10 |
Día 6 |
0,98 [0,87; 1,10] |
1,01 [0,91; 1,11] |
| Raloxifeno 60 mg 1 vez al día del día 1 al 3 |
1200 mg 2 veces al día el día 1, 800 mg 2 veces al día el día 2, 800 mg 1 vez al día el día 3 |
17 |
Día 1 |
1,00 [0,90; 1,10] |
1,03 [0,95; 1,12] |
| Día 3 |
0,90 [0,81; 0,99] |
0,85 [0,79; 0,93] |
|||
| Hidralazina 5 mg 1 vez al día el día 1 y el día 5 |
1200 mg/400 mg el día 1, 400 mg 2 veces al día del día 2 al 4, 400 mg 1 vez al día el día 5 |
14 |
Día 1 |
0,99 [0,92; 1,06] |
0,99 [0,92; 1,07] |
| Día 5 |
0,96 [0,89; 1,04] |
1,04 [0,96; 1,12] |
Efecto del favipiravir sobre la farmacocinética de los medicamentos concomitantes
| Medicamento concomitante y dosis |
Dosificación de favipiravir |
n |
Momento de la dosificación |
Relación de parámetros para medicamentos concomitantes (90 % IC) (tratamiento conjunto/tratamiento aislado) |
|
| Cmax |
AUC |
||||
| Teofilina 200 mg dos veces al día del día 1 al 9, 200 mg una vez al día el día 10 |
600 mg dos veces al día el día 6, 600 mg una vez al día del día 7 al 10 |
10 |
Día 7 |
0,93 [0,85; 1,01] |
0,92 [0,87; 0,97] |
| Día 10 |
0,99 [0,94; 1,04] |
0,97 [0,91; 1,03] |
|||
| Oseltamivir 75 mg dos veces al día del día 1 al 5, 75 mg una vez al día el día 6 |
600 mg dos veces al día el día 5, 600 mg una vez al día el día 6 |
10 |
Día 6 |
1,10 [1,06; 1,15] |
1,14 [1,10; 1,18] |
| Paracetamol 650 mg una vez al día el día 1 y el día 5 |
1200 mg dos veces al día el día 1, 800 mg dos veces al día del día 2 al 4, 800 mg una vez al día el día 5 |
28 |
Día 1 |
1,03 [0,93; 1,14] |
1,16 [1,08; 1,25] |
| Día 5 |
1,08 [0,96; 1,22] |
1,14 [1,04; 1,26] |
|||
| Noretindrona/etinilestradiol 1 mg/0,035 mg una vez al día del día 1 al 4 |
1200 mg dos veces al día el día 1, 800 mg dos veces al día del día 2 al 4, 800 mg una vez al día el día 5 |
25 |
Día 12 |
1,23 [1,16; 1,30] |
1,47 [1,42; 1,52] |
| Día 12 |
1,48 [1,42; 1,54] |
1,43 [1,39; 1,47] |
|||
| Repaglinida 0,5 mg una vez al día el día 13 |
1200 mg dos veces al día el día 1, 800 mg dos veces al día del día 2 al 4, 800 mg una vez al día el día 5 |
17 |
Día 13 |
1,28 [1,16; 1,41] |
1,52 [1,37; 1,68] |
| Hidralazina 5 mg una vez al día el día 1 y el día 5 |
1200 mg/400 mg el día 1, 400 mg dos veces al día del día 2 al 4, 400 mg una vez al día el día 5 |
14 |
Día 1 |
0,73 [0,67; 0,81] |
0,87 [0,78; 0,97] |
| Día 5 |
0,79 [0,71; 0,88] |
0,91 [0,82; 1,01] |
|||
Características de uso.
La administración del medicamento FAVIPIRAVIR-MIKROKIM para el tratamiento del virus de la gripe solo es posible en ausencia de efecto de otro tratamiento y cuando el beneficio esperado para el paciente supere el riesgo.
Advertencia
Dado que en estudios en animales se han observado casos de muerte embrionaria en etapas tempranas del desarrollo y efectos teratogénicos, no se debe administrar el medicamento a mujeres con embarazo confirmado o sospechado (véase la sección «Contraindicaciones» y «Uso durante el embarazo o la lactancia»).
Al administrar el medicamento a mujeres en edad fértil, antes del inicio del tratamiento se debe confirmar un resultado negativo en la prueba de embarazo. Se debe informar detalladamente a las mujeres sobre los riesgos asociados con el uso de este medicamento y se debe recomendar encarecidamente el uso de métodos anticonceptivos altamente eficaces por parte de la mujer y su pareja durante el tratamiento con este fármaco y durante 7 días posteriores a la finalización de su uso (véase la sección «Uso durante el embarazo o la lactancia»). Si durante el tratamiento surge una sospecha de embarazo, se debe recomendar a la mujer que suspenda inmediatamente la toma del medicamento y consulte con su médico.
El favipiravir penetra en el semen. Al administrar el medicamento a pacientes de sexo masculino, se debe informar detalladamente sobre los riesgos asociados con el tratamiento y se debe recomendar encarecidamente el uso de métodos anticonceptivos altamente eficaces durante las relaciones sexuales durante el tratamiento y durante 7 días posteriores a su finalización (los hombres deben usar condones). Además, a los pacientes de sexo masculino se les debe informar que no deben tener relaciones sexuales con mujeres embarazadas (véanse las secciones «Uso durante el embarazo o la lactancia» y «Farmacocinética»).
Antes del inicio del tratamiento, se debe informar detalladamente sobre la eficacia del medicamento y sus riesgos (incluido el riesgo de efectos sobre el feto).
Antes de iniciar el tratamiento con el medicamento, se debe evaluar cuidadosamente la conveniencia de su uso.
Precauciones
FAVIPIRAVIR-MIKROKIM es un medicamento cuyo uso solo debe considerarse en caso de brotes de nuevas infecciones o reaparición de infecciones causadas por el virus de la gripe, cuando otros medicamentos antivirales sean ineficaces o insuficientemente eficaces. Durante el tratamiento con este medicamento, se debe consultar la información más reciente y administrar el fármaco únicamente a pacientes adecuados.
El favipiravir no es eficaz contra infecciones bacterianas.
El favipiravir no debe administrarse a niños (véase la sección «Niños»).
Independientemente de la vía de administración o del tipo de agentes antivirales contra la gripe, se han registrado casos de comportamiento anormal en pacientes con infección por el virus de la gripe (véase la sección «Reacciones adversas»). Para prevenir accidentes, como caídas debido a comportamiento inusual, como medida preventiva, se debe instruir a los pacientes o sus cuidadores sobre lo siguiente:
- puede presentarse comportamiento anormal,
- cuando los pacientes sean tratados en casa, los cuidadores u otras personas deben tomar medidas preventivas contra accidentes, como caídas, durante al menos 2 días después del inicio de la fiebre.
Las formas graves de comportamiento anormal que han provocado accidentes por caídas se han observado con mayor frecuencia en niños en edad escolar y menores de edad. Se sabe que estos síntomas aparecen con mayor probabilidad durante los primeros 2 días tras el inicio de la fiebre.
Se debe observar cuidadosamente a los pacientes y, si se detectan alteraciones, se debe suspender el tratamiento y tomar las medidas adecuadas.
La infección causada por el virus de la gripe puede complicarse con infecciones bacterianas o presentarse con síntomas que pueden confundirse fácilmente con los de la gripe. En caso de infección bacteriana o sospecha de infección bacteriana, se deben tomar medidas adecuadas, como la administración de antibacterianos.
Uso durante el embarazo o la lactancia.
Embarazo
Está contraindicado el uso del medicamento FAVIPIRAVIR-MIKROKIM en mujeres con embarazo conocido o sospechado. En estudios en animales, con niveles de exposición similares o inferiores a la exposición clínica, se observaron muerte embrionaria en etapas tempranas (en ratas) y efectos teratogénicos (en monos, ratones, ratas y conejos).
Lactancia
Al administrar el medicamento FAVIPIRAVIR-MIKROKIM a mujeres que amamantan, se debe suspender la lactancia. Durante los estudios se determinó que el metabolito principal del favipiravir, la forma hidroxilada, penetra en la leche materna.
Dado que en estudios en animales se han observado casos de muerte embrionaria en etapas tempranas del desarrollo y efectos teratogénicos, no se debe administrar favipiravir a mujeres con embarazo confirmado o sospechado (véase la sección «Contraindicaciones»).
Al administrar favipiravir a mujeres en edad fértil, antes del inicio del tratamiento se debe confirmar un resultado negativo en la prueba de embarazo. Se debe informar detalladamente a las mujeres sobre los riesgos asociados con el uso de este medicamento y se debe recomendar encarecidamente el uso de métodos anticonceptivos altamente eficaces por parte de la mujer y su pareja durante el tratamiento con este fármaco y durante 7 días posteriores a la finalización de su uso. Si durante el tratamiento surge una sospecha de embarazo, se debe recomendar a la mujer que suspenda inmediatamente la toma del medicamento y consulte con su médico.
El favipiravir penetra en el semen. Al administrar el medicamento a pacientes de sexo masculino, se debe informar detalladamente sobre los riesgos asociados con el tratamiento y se debe recomendar encarecidamente el uso de métodos anticonceptivos altamente eficaces durante las relaciones sexuales durante el tratamiento y durante 7 días posteriores a su finalización (los hombres deben usar condones). Además, a los pacientes de sexo masculino se les debe informar que no deben tener relaciones sexuales con mujeres embarazadas (véase la sección «Farmacocinética»).
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar mecanismos.
No se dispone de información sobre el efecto del favipiravir en la velocidad de reacción al conducir automóviles o al trabajar con otros mecanismos.
Vía de administración y dosis.
La dosis habitual de favipiravir para adultos es de 1600 mg: se administra por vía oral dos veces al día en el primer día, seguido de 600 mg por vía oral dos veces al día durante los siguientes 4 días.
La duración total del tratamiento debe ser de 5 días.
El tratamiento debe iniciarse inmediatamente tras la aparición de los síntomas de gripe.
Grupos de pacientes especiales
Pacientes de edad avanzada (> 65 años)
Dado que en las personas de edad avanzada las funciones fisiológicas suelen estar disminuidas, se debe administrar el medicamento FAVIPIRAVIR-MIKROKHIM con precaución, controlando el estado general del paciente.
Pacientes pediátricos (< 18 años)
El medicamento no está indicado para su uso en niños.
Alteración de la función hepática
En pacientes con alteración hepática leve y moderada (clases A y B según la clasificación de Child-Pugh, 6 pacientes por cada clase), tras la administración oral de favipiravir a dosis de 1200 mg dos veces al día el primer día y luego 800 mg dos veces al día durante los siguientes 4 días, en comparación con voluntarios adultos sanos, los valores de Cmax y AUC el día 5 fueron aproximadamente 1,6 y 1,7 veces más altos, respectivamente, en pacientes con alteración hepática leve, y 1,4 y 1,8 veces más altos en pacientes con alteración hepática moderada.
En pacientes con alteración hepática grave (clase C según la clasificación de Child-Pugh, 4 individuos), tras la administración oral de favipiravir a dosis de 800 mg dos veces al día el primer día y luego 400 mg dos veces al día durante 2 días, en comparación con voluntarios adultos sanos, los valores de Cmax y AUC el día 3 fueron aproximadamente 2,1 y 6,3 veces más altos, respectivamente.
Situaciones que requieren precaución
El medicamento debe administrarse con precaución a pacientes con gota activa o antecedentes de gota, así como a pacientes con hiperuricemia (en estos pacientes puede aumentar el nivel de ácido úrico en sangre, por lo que los síntomas podrían agravarse).
Niños
El medicamento no se utiliza en la población pediátrica.
Sobredosis
No se dispone de información sobre sobredosis con favipiravir.
Reacciones adversas.
Favipiravir no se ha administrado a la dosis aprobada. En estudios clínicos japoneses y en un estudio global de fase III (estudios realizados con niveles de dosis inferiores a los aprobados), se observaron reacciones adversas en 100 de 501 pacientes (19,96 %) y se evaluaron desde el punto de vista de la seguridad (incluyendo valores anormales en pruebas de laboratorio).
Las reacciones adversas principales incluyeron: aumento de los niveles de ácido úrico en sangre (en 24 pacientes (4,79 %)), diarrea (en 24 pacientes (4,79 %)), disminución del número de neutrófilos (en 9 pacientes (1,80 %)), elevación de AST (GOT) (en 9 pacientes (1,80 %)), aumento de ALT (GPT) (en 8 pacientes (1,60 %)).
Reacciones adversas clínicamente significativas
Aunque la relación causal es desconocida, se han notificado síntomas psiconeuróticos, como comportamiento anormal, tras la administración de medicamentos contra el virus de la gripe, incluyendo favipiravir (véase la sección «Precauciones de uso»).
Reacciones adversas clínicamente significativas (observadas con medicamentos análogos)
Las siguientes reacciones adversas clínicamente significativas se han registrado con otros medicamentos utilizados contra el virus de la gripe.
Del sistema inmunitario: shock, anafilaxia.
Del sistema respiratorio, órganos del tórax y mediastino: neumonía.
Del sistema hepatobiliar: hepatitis fulminante, alteración de la función hepática, ictericia.
De la piel y tejido subcutáneo: necrólisis epidérmica tóxica (NET), síndrome de Stevens-Johnson.
De los riñones y sistema urinario: insuficiencia renal aguda.
Del sistema sanguíneo y sistema linfático: disminución del número de leucocitos, neutrófilos y plaquetas.
Del estado psíquico: alteración de la conciencia, delirio, alucinaciones, manía, convulsiones.
Del tracto gastrointestinal: colitis hemorrágica.
Debe mantenerse una vigilancia constante del paciente y, en caso de aparición de reacciones adversas, debe interrumpirse el tratamiento y adoptarse las medidas adecuadas.
Otras reacciones adversas observadas en estudios clínicos japoneses y en el estudio clínico global de fase III (estudios realizados con niveles de dosis inferiores a los aprobados)
| Clasificación de órganos del sistema MedDRA |
Reacciones adversas |
||
| ≥ 1 % |
0,5 – < 1 % |
< 0,5 % |
|
| Hipersensibilidad |
|
Erupción cutánea |
Exantema, prurito |
| Trastornos del sistema hepatobiliar |
Aumento de AST (GOT), ALT (GPT), γ-GT |
|
Aumento de fosfatasas alcalinas y bilirrubina en sangre |
| Trastornos del tubo digestivo |
Diarrea (4,79 %) |
Náuseas, vómitos, dolor abdominal |
Molestias abdominales, úlcera duodenal, hematocacía, gastritis |
| Trastornos de la sangre y del sistema linfático |
Disminución del número de neutrófilos y leucocitos |
|
Aumento del número de leucocitos y monocitos, disminución del número de reticulocitos |
| Trastornos del metabolismo y nutrición |
Aumento de ácido úrico (4,79 %) y triglicéridos en sangre |
Glucosuria |
Disminución del potasio en sangre |
| Trastornos del sistema respiratorio, del tórax y mediastino |
|
|
Asma bronquial, dolor orofaringeo, rinitis, nasofaringitis |
| Otros |
|
|
Aumento de CK (CPK), presencia de sangre en orina, pólipo de amígdalas, pigmentación, disgeusia, equimosis, visión borrosa, dolor ocular, mareo, extrasístole supraventricular |
Ante la aparición de las reacciones adversas anteriormente mencionadas, se deben tomar las medidas adecuadas de acuerdo con la sintomatología presente.
Plazo de validez.
1,5 años.
Condiciones de almacenamiento.
Conservar a una temperatura no superior a 25 °C en el envase original.
Conservar en un lugar inaccesible para los niños.
Envase.
10 comprimidos por blíster, 4 blísteres por estuche de cartón, o 10 comprimidos por blíster, 10 blísteres por estuche de cartón.
O bien, 40 comprimidos en un frasco, 1 frasco por estuche de cartón, o 100 comprimidos en un frasco, 1 frasco por estuche de cartón.
Categoría de dispensación.
Bajo receta médica.
Fabricante.
NVF «MIKROKHIM» S.L.
Domicilio del fabricante y dirección del lugar de ejercicio de su actividad.
Ucrania, 01013, Kiev, calle Budindustrii, 5.
Solicitante.
NVF «MIKROKHIM» S.L.
Domicilio del solicitante.
Ucrania, 01013, Kiev, calle Budindustrii, 5.