Esperoct
UcraniaContenido
INSTRUCCIONES PARA USO MÉDICO DEL MEDICAMENTO ESPEROC (Esperoct)
Composición:
Principio activo: turcoctocog alfa pegol;
500 UI
Un frasco con polvo contiene una cantidad nominal de 500 UI de turcoctocog alfa pegol*.
Después de la reconstitución del medicamento, 1 ml de solución contiene aproximadamente 125 UI de turcoctocog alfa pegol.
1000 UI
Un frasco con polvo contiene una cantidad nominal de 1000 UI de turcoctocog alfa pegol*.
Después de la reconstitución del medicamento, 1 ml de solución contiene aproximadamente 250 UI de turcoctocog alfa pegol.
1500 UI
Un frasco con polvo contiene una cantidad nominal de 1500 UI de turcoctocog alfa pegol*.
Después de la reconstitución del medicamento, 1 ml de solución contiene aproximadamente 375 UI de turcoctocog alfa pegol.
2000 UI
Un frasco con polvo contiene una cantidad nominal de 2000 UI de turcoctocog alfa pegol*.
Después de la reconstitución del medicamento, 1 ml de solución contiene aproximadamente 500 UI de turcoctocog alfa pegol.
3000 UI
Un frasco con polvo contiene una cantidad nominal de 3000 UI de turcoctocog alfa pegol*.
Después de la reconstitución del medicamento, 1 ml de solución contiene aproximadamente 750 UI de turcoctocog alfa pegol.
La actividad (en unidades internacionales, UI) se determina mediante un método cromogénico según la Farmacopea Europea. La actividad específica del turcoctocog alfa pegol es aproximadamente de 9500 UI/mg de proteína.
El principio activo turcoctocog alfa pegol es un conjugado covalente de la proteína turcoctocog alfa* con polietilenglicol (PEG) de tamaño 40 kDa.
* Factor VIII humano producido mediante tecnología de ADN recombinante en una línea celular de ovario de hámster chino, sin que se utilicen aditivos de origen humano o animal en esta línea celular, ni durante la purificación, conjugación o fabricación de los componentes del medicamento Esperoct.
Excipientes:
polvo: L-histidina; sacarosa; polisorbato 80; cloruro de sodio; L-metionina; cloruro de calcio, dihidrato; hidróxido de sodio (para ajuste del pH); ácido clorhídrico (para ajuste del pH);
disolvente: cloruro de sodio; agua para inyección.
Forma farmacéutica. Polvo y disolvente para solución inyectable.
Principales propiedades físico-químicas: el polvo liofilizado presenta un aspecto de liofilizado de color blanco a prácticamente blanco.
El disolvente es transparente e incoloro; pH 6,9; osmolaridad 590 mOsmol/kg.
Grupo farmacoterapéutico. Agentes hemostáticos. Factor de coagulación VIII.
Código ATC B02B D02.
Propiedades farmacológicas.
Farmacodinámica.
Mecanismo de acción
Turcoctocog alfa pegol es un factor VIII recombinante humano purificado (rFVIII) al que se ha conjugado una molécula de polietilenglicol (PEG) de 40 kDa. El PEG está unido a un glicano O-ligado en el dominio B truncado del rFVIII (turcoctocog alfa). El mecanismo de acción de turcoctocog alfa pegol se basa en la sustitución del factor VIII deficiente o ausente en pacientes con hemofilia A.
Cuando turcoctocog alfa pegol es activado por trombina en el sitio de lesión, el dominio B que contiene el fragmento de PEG y la región a3 se separan, dando lugar a un factor VIII recombinante activado (rFVIIIa) estructuralmente similar al factor VIIIa nativo.
El complejo del factor VIII con el factor de von Willebrand está compuesto por dos moléculas (factor VIII y factor de von Willebrand), cada una con funciones fisiológicas distintas. Tras la administración del medicamento a un paciente con hemofilia, el factor VIII se une al factor de von Willebrand en la circulación. El factor VIII activado actúa como cofactor del factor IX activado, acelerando la conversión del factor X en factor X activado.
El factor X activado convierte la protrombina en trombina. Posteriormente, la trombina convierte el fibrinógeno en fibrina, permitiendo así la formación del coágulo. La hemofilia A es un trastorno hereditario ligado al cromosoma X del sistema de coagulación sanguínea debido a la disminución de los niveles de factor VIII:C, y provoca hemorragias abundantes en articulaciones, músculos o órganos internos, ya sea de forma espontánea o tras traumatismos accidentales o intervenciones quirúrgicas. La terapia sustitutiva con factor VIII eleva los niveles de este factor en el plasma sanguíneo, permitiendo así una corrección temporal de la deficiencia y contrarrestando la tendencia a la hemorragia.
Eficacia clínica en la profilaxis y tratamiento de episodios hemorrágicos
La eficacia clínica del medicamento Esperoct en la profilaxis y el tratamiento de hemorragias se evaluó en siete estudios clínicos prospectivos multicéntricos. Todos los pacientes tenían hemofilia A grave.
Debe tenerse en cuenta que no se deben comparar directamente las tasas anuales de hemorragia (TAH) obtenidas con diferentes concentrados de factores ni entre distintos estudios clínicos.
Profilaxis en adultos/niños de 12 años o más
La eficacia del uso de Esperoct para la profilaxis y el tratamiento de hemorragias se evaluó en un estudio abierto, no controlado, con adultos y niños de 12 años o más con hemofilia A grave. El efecto profiláctico de Esperoct se demostró con una dosis de 50 UI/kg administrada cada 4 días o cada 3-4 días (dos veces por semana) en 175 pacientes. La tasa media anual de hemorragia (TAH) en adultos y niños de 12 años o más que recibieron Esperoct fue de 1,18 (rango intercuartílico [RI]: 0,00; 4,25), siendo la TAH para episodios espontáneos de 0,00 (RI: 0,00; 1,82), para traumatismos de 0,00 (RI: 0,00; 1,74) y para hemorragias articulares de 0,85 (RI: 0,00; 2,84). Al incluir supuestos condicionales (para reemplazar datos faltantes de pacientes que abandonaron el estudio), la tasa media calculada de TAH para todos los episodios hemorrágicos fue de 3,70 (IC del 95 %: 2,94; 4,66). De los 175 adultos/niños de 12 años o más en profilaxis, 70 (40 %) no presentaron ninguna hemorragia. La dosis media anual de administración del medicamento con fines profilácticos fue de 4 641 UI/kg.
Los adultos/niños de 12 años o más con baja frecuencia de hemorragias (0-2 episodios en los últimos 6 meses) y que habían recibido al menos 50 dosis de Esperoct, pudieron ser aleatorizados a regímenes profilácticos con el medicamento cada 7 días (75 UI/kg cada 7 días) o cada 4 días (50 UI/kg cada 4 días). En total, 55 de los 120 pacientes que cumplían los criterios aceptaron ser aleatorizados (17 al grupo de dosis cada 4 días y 38 al grupo de dosis cada 7 días). La TAH en los pacientes aleatorizados fue de 1,77 (0,59; 5,32) con dosificación cada 4 días y de 3,57 (2,13; 6,00) con profilaxis semanal. Durante la fase aleatorizada del estudio, nueve de estos pacientes regresaron a la profilaxis cada 4 días. En total, considerando todas las fases del estudio de extensión, 31 de los 61 pacientes que recibían profilaxis cada 7 días regresaron al régimen de tratamiento cada 4 días.
Profilaxis en pacientes previamente tratados (menores de 12 años)
La eficacia y seguridad del uso de Esperoct para la profilaxis y el tratamiento a demanda de episodios hemorrágicos se evaluaron en un estudio abierto, unicéntrico y no controlado con 68 niños menores de 12 años con hemofilia A grave.
El efecto profiláctico de Esperoct se demostró con una dosis profiláctica de 64,7 UI/kg administrada dos veces por semana. La mediana y la media calculada anual de episodios hemorrágicos en niños menores de 12 años que recibieron Esperoct dos veces por semana fueron de 1,95 y 2,13, respectivamente (IC del 95 %: 1,48; 3,06); para episodios espontáneos, la TAH fue de 0,00 y 0,58 (IC del 95 %: 0,24; 1,40); para hemorragias por traumatismo, de 0,00 y 1,52 (IC del 95 %: 1,07; 2,17); y para hemorragias articulares, de 0,00 y 1,03 (IC del 95 %: 0,59; 1,81), respectivamente. En el grupo de 68 niños menores de 12 años, 29 pacientes (42,6 %) que recibieron Esperoct de forma profiláctica no presentaron ninguna hemorragia.
La necesidad media anual de medicamento para profilaxis fue de 6 475 UI/kg.
Debido a la larga duración del estudio, varios pacientes salieron del grupo de edad al que inicialmente habían sido asignados: algunos pacientes < 6 años también entraron en la categoría de 6-11 años, y algunos pacientes del grupo de 6-11 años pasaron a la categoría de adolescentes. Los resultados principales de la evaluación de eficacia en pacientes menores de 12 años, obtenidos en las fases principal y de extensión del estudio, se resumen en la Tabla 1.
Tabla 1. Frecuencia anual de hemorragias (TAH) en el estudio con pacientes pediátricos previamente tratados según grupos de edad actuales (fases principal y de extensión del estudio) – población total de análisis
| Fase principal |
Fase prolongada |
|||
| Edad del paciente* |
0–5 años (N = 34) |
6–11 años (N = 34) |
0–5 años (N = 27) |
6–11 años (N = 53) |
| Número de hemorragias |
30 |
32 |
41 |
134 |
| Período principal de tratamiento (años) |
0,46 |
0,51 |
4,79 |
4,86 |
| Frecuencia anual total de hemorragias (TFR) |
||||
| Media de Poisson |
1,94 |
1,84 |
0,32 |
0,52 |
| Mediana |
1,94 |
1,94 |
0,22 |
0,21 |
* Algunos pacientes pertenecían a dos grupos de edad.
Prevención en pacientes previamente no tratados (PUPs) (niños menores de 6 años)
La eficacia y la seguridad del uso del medicamento Esperoct se evaluaron en un estudio internacional, abierto y no aleatorizado de Fase 3. La prevención previa (profilaxis opcional con tratamiento a demanda de episodios de hemorragia y/o dosificación de 60 U/mg con intervalos superiores a una semana, hasta que el paciente alcanzara 20 días de exposición (DE) o la edad de 24 meses) y la prevención de hemorragias se evaluaron en 81 pacientes previamente no tratados, menores de 6 años, con hemofilia A grave. De los 81 pacientes, 55 comenzaron con la profilaxis previa, de los cuales 42 posteriormente pasaron a la profilaxis. En total, 69 pacientes recibieron profilaxis con una dosis profiláctica media de 68,9 U/kg de peso corporal administrada dos veces por semana.
El efecto profiláctico del medicamento Esperoct en niños previamente no tratados menores de 6 años con hemofilia A grave se demostró mediante una frecuencia anual mediana de episodios hemorrágicos de 1,35 y una frecuencia media calculada de 1,76, respectivamente (IC del 95 %: 1,26; 2,46).
La necesidad anual media para los 69 pacientes previamente no tratados que recibieron profilaxis fue de 5.395 U/kg.
Los principales resultados de la evaluación de la eficacia en pacientes previamente no tratados que recibieron profilaxis, desglosados por fase principal y fase prolongada del estudio, se resumen en la Tabla 2.
Tabla 2. Frecuencia anual de hemorragias (FAH) en el estudio con participación de pacientes pediátricos previamente no tratados (fases principal y prolongada): población total de análisis
| Fase principal |
Fase prolongada |
|
| Número de hemorragias |
124 |
223 |
| Período principal de tratamiento (años) |
0,60 |
2,83 |
| Frecuencia total de hemorragias por año (FTAH) |
||
| Media de Poisson (IC del 95 %) |
2,98 (2,16; 4,10) |
1,43 (0,98; 2,10) |
| Mediana |
2,49 (0,00; 5,22) |
0,73 (0,00; 2,57) |
Durante el estudio se notificaron 56 eventos adversos en 43 de 81 pacientes y un total de 80 reacciones adversas graves en 48 pacientes tras la administración del medicamento Esperoct.
En 31 de 59 pacientes previamente no tratados sin inhibidores, se observó una reducción transitoria en la recuperación incremental (IR) del factor VIII tras la administración de Esperoct. Hubo 17 pacientes previamente no tratados con mediciones consecutivas de IR reducida, todos ellos presentaron anticuerpos IgG anti-PEG. No puede descartarse una relación entre los anticuerpos anti-PEG y la baja IR.
Eficacia clínica de Esperoct en el tratamiento de episodios hemorrágicos y en su uso a demanda
La eficacia clínica de Esperoct en el tratamiento de episodios hemorrágicos fue demostrada en todos los pacientes previamente tratados de todas las categorías de edad.
La mayoría de los episodios hemorrágicos tratados con Esperoct fueron de gravedad leve a moderada.
La tasa general de éxito de la terapia hemostática para el tratamiento de hemorragias en pacientes previamente tratados fue del 84,4 %.
Las tasas de éxito de la terapia hemostática en los grupos de edad de pacientes previamente tratados fueron del 89,4 % (0-5 años), 82,6 % (6-11 años), 78,9 % (12-17 años) y 84,9 % (≥ 18 años), respectivamente; el 94,2 % se trataron con éxito, con resolución de la hemorragia tras la administración de 1-2 inyecciones.
La eficacia de Esperoct en el tratamiento de episodios hemorrágicos fue demostrada en pacientes previamente no tratados menores de 6 años. La tasa general de éxito de la terapia hemostática fue del 91,9 %; el 93,3 % se trataron con éxito, con resolución de la hemorragia tras la administración de 1-2 inyecciones.
En el estudio principal, 12 pacientes de 18 años o más eligieron continuar con el tratamiento a demanda. En estos pacientes, 1.270 hemorragias fueron tratadas con una dosis media de 37,5 UI/kg (20-75 UI/kg). Tras la administración de 1-2 inyecciones de Esperoct, el 97 % de las hemorragias se detuvieron.
Eficacia clínica de Esperoct en procedimientos quirúrgicos
El efecto hemostático de Esperoct en procedimientos quirúrgicos fue evaluado en cuatro estudios, uno de los cuales fue un estudio específico durante intervenciones quirúrgicas.
En el estudio quirúrgico específico se analizaron 49 intervenciones quirúrgicas mayores en 35 pacientes previamente tratados de edad adolescente y adulta. El día de la cirugía, los pacientes recibieron una dosis media preoperatoria de 55,7 UI/kg (rango: 27,2-86,2 UI/kg), y una dosis media posoperatoria de 30,7 UI/kg (rango: 10,1-58,8 UI/kg). La tasa general de éxito de la terapia hemostática con Esperoct durante intervenciones quirúrgicas mayores fue del 95,9 %, y la eficacia del hemostático fue evaluada como excelente o buena en 47 de las 49 intervenciones realizadas.
En dos estudios con pacientes pediátricos previamente tratados (menores de 12 años), 24 pacientes experimentaron 46 procedimientos quirúrgicos, de los cuales solo uno fue una cirugía mayor con respuesta hemostática exitosa. Las cirugías menores en estos pacientes no provocaron complicaciones, aunque durante estas intervenciones no se controló la eficacia del hemostático ni los niveles de factor VIII. En 26 pacientes pediátricos previamente no tratados (menores de 6 años) en un estudio con pacientes previamente no tratados, se observó un efecto hemostático exitoso en las 4 cirugías mayores y en 25 de las 30 cirugías menores. Esperoct se administró de acuerdo con las recomendaciones de dosificación.
Farmacocinética
En total, se evaluaron 129 perfiles farmacocinéticos (PK) obtenidos tras una dosis única de Esperoct en 86 pacientes (incluyendo 24 pacientes pediátricos de 0 a 12 años).
Todos los estudios de farmacocinética con Esperoct se realizaron en pacientes con hemofilia A grave (niveles de factor VIII < 1 %) previamente tratados. Los pacientes recibieron una dosis única de 50 UI/kg, y las muestras de sangre se recogieron antes de la administración y en múltiples momentos durante las 96 horas siguientes.
En pacientes adultos, la semivida de eliminación de Esperoct fue 1,6 veces más larga que la de los fármacos de factor VIII no modificado disponibles.
Parámetros farmacocinéticos
Se evaluaron 108 perfiles farmacocinéticos obtenidos tras una dosis única de 50 UI/kg de Esperoct en 69 pacientes. Los parámetros farmacocinéticos tras una dosis única fueron comparables entre niños pequeños (0-6 años) y niños de edad media (6-12 años), así como entre adolescentes (12-17 años) y adultos (≥ 18 años).
Como se esperaba, en niños menores de 12 años, en comparación con adultos y adolescentes de 12 años o más, la recuperación incremental (IR) fue menor, mientras que la depuración corregida por peso corporal fue mayor. En general, se observó una tendencia al aumento de la recuperación incremental (IR) y a la disminución de la depuración (ml/h/kg) con la edad. Esto concuerda con el hecho de que en niños menores de 12 años el volumen de distribución por kilogramo de peso corporal es mayor que en adultos (tabla 1).
Los parámetros farmacocinéticos de Esperoct tras una dosis única, determinados tras 28 semanas de tratamiento profiláctico, fueron similares a los parámetros farmacocinéticos iniciales.
En la tabla 3 se muestran los parámetros farmacocinéticos de Esperoct tras una dosis única.
Tabla 3. Parámetros farmacocinéticos de Esperoct 50 UI/kg tras una dosis única en pacientes previamente tratados, determinados mediante método cromogénico [media geométrica (CV % – coeficiente de variación)]
| Parámetro PK, |
Pacientes de 0 a 6 años de edad (N = 13) |
Pacientes de 6 a 12 años de edad (N = 11) |
Pacientes de 12 a 18 años de edad (N = 3) |
Pacientes de 18 años de edad (N = 42) |
| Número de perfiles PK |
13 |
11 |
5 |
79 |
| IR (MO/dl) a (MO/kg)a |
1,80 (29) |
1,99 (25) |
2,79 (12) |
2,63 (22) |
| Actividad máxima del factor VIII (MO/dl)a |
101,2 (28) |
119,6 (25) |
133,2 (9) |
134,4 (23) |
| t1/2 (horas) |
13,6 (20) |
14,2 (26) |
15,8 (43) |
19,9 (34) |
| AUCinf (MO*hora/dl) |
2 147 (47) |
2 503 (42) |
3 100 (44) |
3 686 (35) |
| CL (ml/hora/kg) |
2,6 (45) |
2,4 (40) |
1,5 (43) |
1,4 (32) |
| Vss (ml/kg) |
44,2 (34) |
41,2 (25) |
33,4 (10) |
37,7 (27) |
| MRT (horas) |
17,0 (22) |
17,3 (31) |
21,7 (45) |
25,2 (29)b |
Abreviaturas: AUC – área bajo la curva farmacocinética, que describe la dinámica temporal de la actividad del factor VIII; t1/2 – tiempo de semivida final de eliminación; MRT – tiempo medio de retención; CL – aclaramiento; Vss – volumen de distribución en estado de equilibrio; IR – recuperación incremental.
a El incremento de recuperación (IR) y la actividad del factor VIII se determinaron a los 30 minutos tras la administración del fármaco en pacientes de 12 años o más, y a los 60 minutos tras la administración (primera toma de muestra para análisis) en niños menores de 12 años.
b Cálculos basados en datos de 67 perfiles.
En un estudio con participación de pacientes pediátricos previamente no tratados, el valor de recuperación incremental (IR) del factor VIII se evaluó en 46 pacientes menores de 6 años tras la primera administración, obteniéndose una media geométrica (CV %) de 1,76 (34) [UI/dl]/[UI/kg]. En 17 de los 59 pacientes previamente no tratados sin inhibidores, se observaron mediciones consecutivas (es decir, 2 o más) de recuperación incremental (IR) del factor VIII temporalmente reducida en un rango de 5–10 dosis efectivas (más información detallada en la sección «Propiedades farmacocinéticas»).
Los valores medios de actividad mínima del factor VIII en pacientes previamente tratados y previamente no tratados, agrupados por edades, se resumen en la Tabla 4.
Tabla 4. Valores medios calculados de actividad mínima del factor VIII en pacientes previamente tratados y previamente no tratados, según grupos de edad
| Actividad mínima del factor VIII |
Pacientes previamente tratados, 60 UI/kg del medicamento Esperoct para profilaxis dos veces por semana |
Pacientes previamente tratados, 50 UI/kg del medicamento Esperoct para profilaxis una vez cada 4 días |
Pacientes previamente no tratados, 60 UI/kg del medicamento Esperoct para profilaxis dos veces por semana |
||
| Grupos de edad al momento de inclusión en el estudio |
0–5 años |
6–11 años |
12–17 años |
≥ 18 años |
0–5 años |
| Número de pacientes incluidos en el análisis |
31 |
34 |
23 |
143 |
81 |
| Número de valores mínimos incluidos en el análisis |
144 |
161 |
112 |
722 |
355 |
| Número de valores mínimos por debajo del LMC |
62 |
43 |
16 |
107 |
128a |
| Resultados del modelo mixtob: |
|||||
| 1,2 |
2,0 |
2,7 |
3,0 |
1,5 |
|
| 0,8; 1,6 |
1,5; 2,7 |
1,8; 4,0 |
2,6; 3,5 |
1,1; 1,9 |
|
LMC – límite inferior de cuantificación
a Los niveles de actividad del factor VIII en plasma por debajo del límite inferior de cuantificación (LMC), establecido en 0,009 UI/ml, se definieron en la mitad del LMC (0,0045 UI/ml).
b Modelo mixto con datos de actividad del factor VIII en plasma transformados logarítmicamente, donde el grupo de edad es un efecto fijo y el paciente es un efecto aleatorio. Se realizó modelización separada para cada tratamiento profiláctico (es decir, para cada frecuencia de dosificación). El nivel mínimo se transformó inversamente a la escala natural.
Solo se incluyeron en el análisis las mediciones obtenidas antes de la administración del medicamento y en estado basal para ese tratamiento profiláctico.
Datos preclínicos de seguridad
Los datos de los estudios preclínicos indican ausencia de peligro particular para los seres humanos, según los resultados de los estudios estándar de seguridad y toxicidad por administración repetida.
Características clínicas.
Indicaciones.
Tratamiento y profilaxis de las hemorragias en pacientes con hemofilia A (déficit hereditario del factor VIII).
Espoklot puede administrarse a todos los grupos de edad.
Contraindicaciones.
Hipersensibilidad al principio activo o a cualquiera de los excipientes del medicamento (ver sección «Composición»).
Reacción alérgica conocida a proteínas de hámster.
Interacción con otros medicamentos y otras formas de interacción.
No se han notificado casos de interacción del factor VIII de coagulación sanguínea humano (ADNr) con otros medicamentos.
Características de uso.
Seguimiento
Con el fin de mejorar el seguimiento de los medicamentos biológicos, el nombre y número de lote del producto administrado deben registrarse claramente.
Hipersensibilidad
Durante el tratamiento con Esperoct pueden presentarse reacciones de hipersensibilidad de tipo alérgico. Este medicamento contiene trazas de proteínas de hámster que pueden provocar reacciones alérgicas en algunos pacientes. Se debe recomendar a los pacientes que interrumpan inmediatamente el tratamiento y consulten a su médico si aparecen síntomas de hipersensibilidad. Los pacientes deben informarse sobre los signos tempranos de hipersensibilidad, incluyendo erupción alérgica, urticaria generalizada, opresión en el pecho, sibilancias, hipotensión y anafilaxia.
En caso de presentarse shock, se debe aplicar el tratamiento estándar para el manejo del shock.
Inhibidores
Una complicación conocida en el tratamiento de personas con hemofilia A es la formación de anticuerpos neutralizantes (inhibidores) contra el factor VIII. Estos inhibidores suelen ser inmunoglobulinas IgG dirigidas contra la actividad procoagulante del factor VIII, cuya concentración se determina cuantitativamente en unidades Bethesda (UB) por mililitro de plasma mediante un método cuantitativo modificado. El riesgo de formación de inhibidores depende de la gravedad de la enfermedad y del nivel de exposición al factor VIII, siendo mayor durante los primeros 50 días de exposición, aunque persiste durante toda la vida, aunque con menor frecuencia.
La relevancia clínica de la formación de inhibidores depende de la titulación del inhibidor: una titulación más baja implica un menor riesgo de respuesta clínica insuficiente que una titulación más alta. En general, todos los pacientes tratados con medicamentos que contienen factor VIII deben vigilarse cuidadosamente mediante observación clínica y análisis de laboratorio adecuados para detectar la aparición de inhibidores. Si no se alcanzan los niveles esperados de actividad del factor VIII en plasma o si no se logra controlar la hemorragia con la dosis adecuada, se debe investigar la posible presencia de inhibidores del factor VIII. En pacientes con niveles altos de inhibidores, el tratamiento con factor VIII puede no ser eficaz, por lo que debe considerarse la posibilidad de utilizar otras alternativas terapéuticas.
El tratamiento de estos pacientes debe estar a cargo de médicos con experiencia en el manejo de la hemofilia y la formación de inhibidores del factor VIII.
Disminución de la actividad del factor VIII en pacientes previamente tratados
En informes poscomercialización se han descrito casos de disminución de la actividad del factor VIII en ausencia de inhibidores detectables del factor VIII en pacientes previamente tratados. Esta disminución de la actividad del factor VIII se observó durante el cambio a Esperoct y, en algunos casos, pudo estar relacionada con la presencia de anticuerpos contra el PEG. Debe considerarse la posibilidad de realizar una evaluación adecuada de la actividad del factor VIII tras el cambio de tratamiento. Para más información, véase la sección «Reacciones adversas».
Eventos cardiovasculares
En pacientes con factores de riesgo cardiovascular preexistentes, el uso de terapia sustitutiva con factor VIII puede aumentar el riesgo cardiovascular.
Complicaciones relacionadas con el uso de catéter
Si es necesario utilizar un dispositivo de acceso venoso central (DAVC), se debe considerar el riesgo de complicaciones asociadas con su uso, incluyendo infecciones locales, bacteriemia y trombosis en el sitio de inserción del catéter.
Niños
Las advertencias y precauciones mencionadas son aplicables tanto en adultos como en niños.
Disminución del incremento de recuperación (IR) del factor VIII de coagulación en pacientes previamente no tratados
En 31 de 59 pacientes previamente no tratados, se observó una disminución del incremento de recuperación (IR) del factor VIII sin presencia de inhibidores del factor VIII detectables en estudios clínicos. De estos, 14 pacientes tuvieron solo una medición aislada de IR bajo, mientras que 17 pacientes tuvieron 2 o más mediciones consecutivas de IR bajo dentro de un intervalo de 5 a 10 dosis efectivas. La disminución del IR fue temporal y, entre los 15 y 70 días efectivos, el IR regresó a valores superiores a 0,6 (UI/dl)/(UI/kg). La disminución del IR se asoció con un aumento en los títulos de anticuerpos contra el polietilenglicol (anti-PEG IgG) en pacientes previamente no tratados, sin formación de inhibidores contra el factor VIII. Mediciones consecutivas de IR bajo podrían asociarse potencialmente con una reducción de la eficacia del medicamento durante ese período. Se recomienda el seguimiento de los pacientes pediátricos, incluyendo el monitoreo de la actividad del factor VIII tras la administración del medicamento. Si, al usar la dosis recomendada de Esperoct, no se controla la hemorragia y/o no se alcanzan los niveles esperados de actividad del factor VIII en ausencia de inhibidores del factor VIII, debe considerarse la posibilidad de ajustar la dosis o la frecuencia de administración, o incluso la interrupción del tratamiento.
Sustancias auxiliares
Este medicamento contiene 30,5 mg de sodio por vial con el producto reconstituido, lo que equivale al 1,5 % de la ingesta diaria máxima recomendada por la OMS de sodio –2,0 g– para un adulto.
Uso durante el embarazo o la lactancia.
No se han realizado estudios de reproducción con factor VIII en animales. La hemofilia A en mujeres es rara, por lo que no existe experiencia clínica sobre el uso de factor VIII durante el embarazo y la lactancia. Por tanto, el factor VIII debe usarse durante el embarazo y la lactancia únicamente cuando esté claramente indicado.
Capacidad para afectar la velocidad de reacción al conducir vehículos o manejar maquinaria.
Esperoct no afecta o afecta de forma insignificante la capacidad para conducir vehículos o manejar maquinaria.
Vía de administración y dosis
El tratamiento debe iniciarse bajo la supervisión de un médico con experiencia en el tratamiento de la hemofilia.
Monitorización durante el tratamiento
Durante el tratamiento se recomienda determinar adecuadamente los niveles de actividad del factor VIII para poder ajustar, si es necesario, el esquema de dosificación del medicamento Esperoct. La respuesta al factor VIII puede variar entre diferentes pacientes, lo que se manifiesta en diferentes valores del período de semivida y del índice de recuperación incremental (IR). En pacientes con peso corporal insuficiente o excesivo, puede ser necesario ajustar la dosis según el peso corporal. En el caso de intervenciones quirúrgicas mayores, es necesario supervisar el proceso de la terapia sustitutiva con factor VIII mediante la determinación de la actividad del factor VIII en el plasma sanguíneo.
La actividad del factor VIII presente en el medicamento Esperoct puede determinarse mediante métodos estándar de análisis cuantitativo del factor VIII: método cromogénico y método de una sola etapa. Al utilizar el método de una sola etapa basado en el tiempo de tromboplastina parcial activado (TTPa) in vitro para determinar la actividad del factor VIII en muestras de sangre de pacientes, los resultados de la actividad del factor VIII en el plasma sanguíneo pueden depender significativamente tanto del tipo de reactivo TTPa como del estándar de referencia utilizado en el análisis cuantitativo.
Al emplear el método de una sola etapa para el análisis de coagulación, se debe evitar el uso de ciertos reactivos a base de sílice, ya que pueden provocar resultados falsamente bajos. Además, pueden existir diferencias significativas entre los resultados obtenidos mediante el método de una sola etapa basado en el TTPa y el método cromogénico, según lo establecido en la Farmacopea Europea. Esto es especialmente importante cuando se cambia de laboratorio analítico y/o de reactivos para el análisis.
Dosificación
La dosis, el intervalo de administración y la duración de la terapia sustitutiva dependen de la gravedad del déficit de factor VIII, del sitio y la severidad de la hemorragia, del nivel objetivo de actividad del factor VIII y del estado clínico del paciente. La cantidad de unidades de factor VIII administrado se expresa en unidades internacionales (UI), referidas al estándar internacional de la OMS para concentrados de medicamentos del factor VIII. La actividad del factor VIII en el plasma sanguíneo se expresa bien en porcentaje (en relación con el nivel normal en plasma humano), bien en unidades internacionales por decilitro (en relación con el estándar internacional para factor VIII en plasma sanguíneo).
Una unidad internacional (UI) de actividad del factor VIII equivale a la cantidad de factor VIII presente en 1 ml de plasma sanguíneo humano.
Tratamiento a demanda y tratamiento de episodios hemorrágicos
El cálculo de la dosis necesaria de factor VIII se basa en resultados empíricos que indican que una unidad internacional (UI) de factor VIII por kilogramo de peso corporal eleva la actividad del factor VIII en el plasma sanguíneo en 2 UI/dl.
La dosis requerida se determina mediante la siguiente fórmula:
Cantidad necesaria de unidades (UI) = peso corporal (kg) × incremento deseado del nivel de factor VIII (%) (UI/dl) × 0,5 (UI/kg por UI/dl).
La cantidad y frecuencia necesarias deben ajustarse siempre según la eficacia clínica observada en cada caso individual y en cada paciente específico.
Las recomendaciones sobre la dosificación de Esperoct para el tratamiento a demanda y para el tratamiento de episodios hemorrágicos se indican en la tabla 5. Los niveles de actividad del factor VIII deben mantenerse en el nivel indicado o por encima de él en el plasma sanguíneo (en UI por decilitro o en % respecto al valor normal). En el tratamiento de hemorragias, puede administrarse una dosis única máxima de 75 UI/kg y una dosis total máxima de 200 UI/kg/24 horas.
Tabla 5. Recomendaciones para el tratamiento de episodios hemorrágicos con Esperoct
| Grado de gravedad del sangrado |
Nivel requerido del factor VIII (UI/dl o % de la normalidad)a |
Frecuencia de dosificación (horas) |
Duración del tratamiento |
| Leve Hemartrosis precoz, hemorragia leve en músculos o en la cavidad bucal |
20–40 |
12–24 |
Hasta que cese la hemorragia |
| Moderado Hemartrosis más extendida, hemorragia muscular, hematoma |
30–60 |
12–24 |
Hasta que cese la hemorragia |
| Hemorragias graves o amenazantes para la vida |
60–100 |
8–24 |
Hasta que el riesgo haya desaparecido |
a La dosis necesaria se determina mediante la siguiente fórmula:
Cantidad necesaria de unidades (UI) = masa corporal (kg) × incremento deseado del nivel del factor VIII (%) (UI/dl) × 0,5 (UI/kg por UI/dl).
Uso en la administración perioperatoria
El nivel de dosis y el intervalo de administración durante intervenciones quirúrgicas dependen del procedimiento quirúrgico empleado y de la práctica local establecida. Se puede administrar una dosis única máxima del medicamento Esperoct de 75 UI/kg y una dosis total máxima de 200 UI/kg/24 horas.
La frecuencia de administración y la duración del tratamiento deben ajustarse siempre individualmente, según la respuesta clínica de cada paciente.
En la tabla 6 se indican recomendaciones generales para la dosificación del medicamento Esperoct en la administración perioperatoria. Debe tenerse en cuenta mantener la actividad del factor VIII en un nivel igual o superior al rango objetivo.
Tabla 6. Recomendaciones para la dosificación del medicamento Esperoct en la administración perioperatoria
| Tipo de procedimiento quirúrgico |
Nivel necesario de factor VIII (UI/dl (%) (UI/dl)a |
Frecuencia de dosificación (horas) |
Duración del tratamiento |
| Intervención quirúrgica menor (incluyendo extracción dental) |
30–60 |
Dentro de la primera hora antes de la intervención quirúrgica. Si es necesario, repetir a las 24 horas. |
Inyección única o repetida cada 24 horas durante al menos un día, hasta alcanzar la recuperación. |
| Intervención quirúrgica mayor |
80–100 (antes y después de la intervención quirúrgica) |
Dentro de la primera hora antes de la intervención quirúrgica, para alcanzar el nivel de actividad del factor VIII dentro del rango objetivo. Repetir cada 8–24 horas para mantener la actividad del factor VIII dentro del rango objetivo. |
Repetir la inyección cada 8–24 horas según sea necesario para lograr un grado adecuado de cicatrización. Considerar la posibilidad de continuar el tratamiento durante otros 7 días para mantener la actividad del factor VIII entre el 30 % y el 60 % (UI/dl). |
a La dosis necesaria se determina mediante la siguiente fórmula:
Cantidad necesaria de unidades (UI) = masa corporal (kg) × aumento deseado del nivel del factor VIII (%) (UI/dl) × 0,5 (UI/kg por UI/dl).
Prevención
La dosis recomendada del medicamento Esperoct en adultos es de 50 UI por kilogramo de peso corporal cada 4 días.
Se puede considerar la posibilidad de ajustar la dosis y los intervalos de administración según los niveles alcanzados de factor VIII y la tendencia individual al sangrado de cada paciente.
Pacientes pediátricos
La dosis recomendada para niños (de 12 años o más) es la misma que para adultos.
La dosis recomendada para profilaxis en niños menores de 12 años es de 65 UI por kg de peso corporal (50–75 UI/kg) dos veces por semana. El ajuste de la dosis y la frecuencia de administración del medicamento dependerá del nivel alcanzado de factor VIII y de la predisposición individual al sangrado.
Para obtener información más detallada sobre pacientes pediátricos, véanse las secciones «Propiedades farmacodinámicas», «Propiedades farmacocinéticas» y «Precauciones de uso especiales».
Vía de administración
El medicamento Esperoct está indicado para administración intravenosa.
Esperoct debe administrarse mediante inyección intravenosa (durante aproximadamente 2 minutos) tras la reconstitución del polvo con 4 ml del disolvente suministrado con el medicamento (cloruro de sodio, 9 mg/ml (0,9 %), solución inyectable).
Las instrucciones para la reconstitución del medicamento antes de su administración se describen más adelante en la sección «Precauciones relativas a manipulación y eliminación».
Precauciones relativas a manipulación y eliminación
Esperoct debe administrarse por vía intravenosa tras la reconstitución del polvo con el disolvente contenido en la jeringa. Tras la reconstitución, la solución debe presentarse como un líquido transparente e incoloro, sin partículas visibles. Antes de la administración, el medicamento reconstituido debe examinarse visualmente para detectar la presencia de partículas sólidas o cambios de color. La solución debe ser transparente e incolora. No utilice la solución si está turbia o contiene sedimentos.
Las instrucciones para la reconstitución del medicamento antes de su administración se describen en la sección «Instrucciones de uso del medicamento Esperoct».
La velocidad de administración debe determinarse según el nivel de comodidad del paciente, durante aproximadamente 2 minutos.
Además, se necesitará un sistema de infusión (aguja-borla con tubo), toallitas estériles con alcohol, gasas y esparadrapo. Estos dispositivos no están incluidos en el envase de Esperoct.
Siempre debe utilizarse técnica aséptica.
Eliminación
Tras la inyección, deseche de forma segura la jeringa con el sistema de infusión, el frasco y el adaptador para frasco.
Cualquier medicamento no utilizado o residuos deben eliminarse de acuerdo con los requisitos locales.
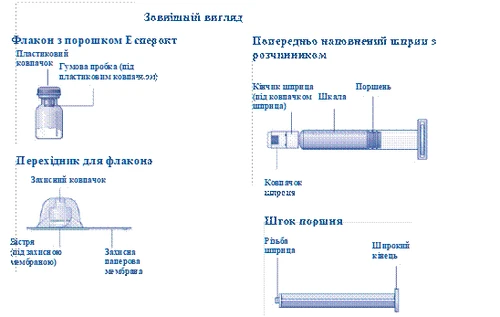
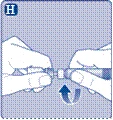
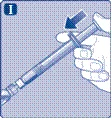
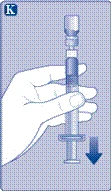
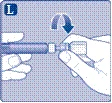
| Instrucciones de uso del medicamento Esperoct Lea cuidadosamente estas instrucciones antes de utilizar el medicamento Esperoct. El medicamento Esperoct se suministra en forma de polvo. Antes de la inyección, debe reconstituirse utilizando el diluyente contenido en la jeringa. Este diluyente es una solución inyectable de cloruro de sodio, 9 mg/ml (0,9 %). El medicamento reconstituido debe administrarse por vía intravenosa (inyección i.v.). El equipo incluido en este envase está diseñado para la reconstitución y administración del medicamento Esperoct. También necesitará:
Estos elementos no están incluidos en el envase de Esperoct. No utilice el equipo si no ha recibido la debida formación de su médico o enfermera. Lávese siempre las manos y asegúrese de disponer de un espacio limpio a su alrededor. Al preparar y administrar un medicamento directamente en una vena, es fundamental utilizar técnicas que garanticen la limpieza y esterilidad (métodos asépticos). El uso incorrecto de estas técnicas puede provocar la entrada de microorganismos e infección de la sangre. No abra el equipo hasta que esté listo para usarlo. No utilice el equipo si ha caído o está dañado. En este caso, utilice un nuevo envase. No utilice el equipo si ha caducado. En este caso, utilice un nuevo envase. La fecha de caducidad está impresa en la caja de cartón, en el frasco, en el adaptador para frasco y en la jeringa precargada. No utilice el equipo si sospecha que está contaminado. En este caso, utilice un nuevo envase. No deseche ninguno de los elementos hasta que no haya completado la inyección de la solución del medicamento reconstituido. Este equipo está diseñado exclusivamente para uso único. |
|
| Contenido El envase contiene:
|
|
|
|
|
No utilice ningún otro método para calentar el frasco y la jeringa precargada. |
|
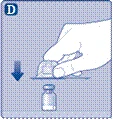
No toque el tapón de goma con los dedos, ya que podría introducir microorganismos. |
|
Si la membrana protectora de papel no está completamente adherida o está rota, no utilice este adaptador para frasco. No saque el adaptador para frasco del protector con los dedos. Si toca la punta del adaptador para frasco, podría introducir microorganismos desde sus dedos. |
|
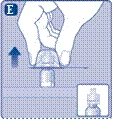
Una vez conectado el adaptador al frasco, no lo retire del frasco. |
|
No desmonte el adaptador del frasco al retirar el protector. |
|
|
|
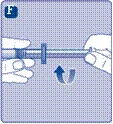
No toque la punta de la jeringa situada debajo de la tapa. Si toca la punta de la jeringa, podría introducir microorganismos desde sus dedos. Si la tapa de plástico está suelta o ausente, no utilice esta jeringa precargada. |
|
|
|
|
|
No agite el frasco, ya que podría provocar formación de espuma.
|
|
| Se recomienda utilizar Esperoct inmediatamente después de la reconstitución. Si no puede utilizar inmediatamente la solución reconstituida de Esperoct, debe usarse:
La solución reconstituida debe conservarse en el frasco. No congele la solución reconstituida ni la guarde en jeringas. Mantenga la solución reconstituida en un lugar protegido de la luz solar directa. Si necesita más de un frasco de Esperoct para obtener la dosis requerida, repita los pasos de la A a la J con frascos adicionales, adaptadores para frascos y jeringas precargadas hasta obtener la cantidad de dosis necesaria. |
|
|
|
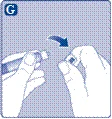
No toque la punta de la jeringa. Si toca la punta de la jeringa, podría introducir microorganismos desde sus dedos. |
|
El medicamento Esperoct ya está listo para la inyección intravenosa.
No mezcle el medicamento Esperoct con otras inyecciones intravenosas ni con otros medicamentos. Administración de Esperoct a través de conectores intravenosos sin aguja. ¡Precaución! La jeringa precargada está fabricada con vidrio y está diseñada para usarse con conexiones estándar tipo Luer-Lok. Algunos conectores sin aguja con espiga interna no son compatibles con esta jeringa precargada. Esta incompatibilidad puede impedir la administración del medicamento y provocar daños en el conector sin aguja. Administración de la solución a través de un dispositivo de acceso venoso central (DAC), como un catéter venoso central o un puerto subcutáneo:
Si es necesario lavar el sistema DAC antes o después de la inyección de Esperoct, utilice solución inyectable de cloruro de sodio a 9 mg/ml (0,9 %). |
|
| Desecho.
No deseche estos materiales con los residuos domésticos habituales. |
|
| No desmonte el equipo antes de desecharlo. No utilice este equipo más de una vez. |
|


Niños
Tratamiento y profilaxis de hemorragias en pacientes con hemofilia A (déficit hereditario del factor VIII).
Esperoct se puede utilizar en todos los grupos de edad.
La información detallada sobre pacientes pediátricos se proporciona en las secciones «Propiedades farmacológicas» (subsecciones «Farmacodinámica» y «Farmacocinética»), «Precauciones de uso», «Vía de administración y dosis».
Sobredosis
No se han notificado casos de síntomas asociados a sobredosis con el factor de coagulación sanguínea VIII recombinante.
Reacciones adversas.
Resumen del perfil de seguridad
Se han observado raramente reacciones de hipersensibilidad o alérgicas (en particular, angioedema, sensación de ardor y picazón en el lugar de la infusión, escalofríos, sofocos, urticaria generalizada, cefalea, erupción alérgica, hipotensión, somnolencia, náuseas, excitación, taquicardia, opresión en el pecho, sensación de hormigueo, vómitos, sibilancias), que en algunos casos pudieron progresar hasta una anafilaxia grave (incluyendo shock).
Muy raramente se ha observado la formación de anticuerpos contra proteínas de hámster, con reacciones de hipersensibilidad correspondientes.
En pacientes con hemofilia A que reciben factor VIII, incluyendo el medicamento Esperoct, pueden desarrollarse anticuerpos neutralizantes (inhibidores). Si se forman tales inhibidores, la falta de respuesta clínica al tratamiento será evidente. En tales casos, se recomienda consultar a un centro especializado en hemofilia.
Lista de reacciones adversas
Los datos sobre la frecuencia de reacciones adversas se obtuvieron de seis estudios clínicos con la participación de 270 pacientes previamente tratados y 81 pacientes previamente no tratados con hemofilia A grave (< 1 % de actividad del factor VIII endógeno) y sin historial de inhibidores del factor VIII, y se presentan en la tabla 7. Las categorías de reacciones adversas registradas a continuación corresponden a la clasificación por sistemas y órganos de MedDRA (por clases de sistemas de órganos y términos de preferencia) indicadas en la tabla 7.
La frecuencia de aparición de reacciones adversas se evaluó según las siguientes categorías: muy frecuentes (≥ 1/10), frecuentes (de ≥ 1/100 a < 1/10), poco frecuentes (de ≥ 1/1000 a < 1/100), raras (de ≥ 1/10000 a < 1/1000), muy raras (< 1/10000); desconocido (no puede estimarse a partir de los datos disponibles). A continuación se indican las reacciones adversas que ocurrieron en pacientes previamente tratados.
Tabla 7. Frecuencia de reacciones adversas registradas durante los estudios clínicos
| Sistemas de órganos según la clasificación |
Términos de uso preferente |
Frecuencia de casos (pacientes previamente tratados) |
Frecuencia de casos (pacientes previamente no tratados) |
| Alteraciones de la sangre y del sistema linfático |
Inhibición del factor VIII * |
Infrecuente |
Muy frecuente** |
| Alteraciones de la piel y del tejido subcutáneo |
Prurito |
Frecuente |
- |
| Eritema |
Frecuente |
Frecuente |
|
| Erupción cutánea |
Frecuente |
Frecuente |
|
| Alteraciones generales y reacciones en el sitio de administración |
Reacciones en el sitio de inyección*** |
Frecuente |
Frecuente |
| Alteraciones del sistema inmunitario |
Hipersensibilidad al medicamento |
- |
Frecuente |
| Hipersensibilidad |
Infrecuente |
- |
|
| Estudios |
Nivel reducido del factor de coagulación sanguínea VIII |
No conocido**** |
- |
* Se definió a un paciente con inhibidor confirmado de factor VIII según el resultado del test inicial de inhibidor ≥ 0,6 unidades Bethesda (UB), confirmado por una segunda muestra obtenida no más tarde de 2 semanas después.
** Incluye pacientes con inhibidor de factor VIII confirmado entre los pacientes en grupo de riesgo (al menos 10 días de tratamiento con el medicamento).
*** Los términos de uso preferente incluyeron reacciones en el sitio de inyección: reacción en el sitio de inyección, hematoma en el sitio de punción vascular, reacción en el sitio de infusión, eritema en el sitio de inyección, erupción cutánea en el sitio de inyección, dolor en el sitio de punción vascular e hinchazón en el sitio de inyección.
**** Basado en informes poscomercialización.
Descripción de reacciones adversas individuales
Formación de inhibidores de factor VIII
Se notificó un caso confirmado de formación de inhibidor de factor VIII durante el uso profiláctico de Esperoct en un paciente de 18 años previamente tratado. El paciente presentaba una inversión del intrón 22 en el gen del factor VIII y un alto riesgo de formación de inhibidores de factor VIII.
No existen evidencias que indiquen un aumento del riesgo de formación de inhibidores de factor VIII con el tratamiento con Esperoct en comparación con otros medicamentos de factor VIII.
Formación de anticuerpos contra el medicamento
Se notificó un caso de formación de anticuerpos persistentes contra el medicamento, que coincidió temporalmente con el caso confirmado de formación de inhibidores de factor VIII (ver sección sobre formación de inhibidores de factor VIII). En tres pacientes, tras la administración de Esperoct, los resultados del test de anticuerpos contra el medicamento fueron temporalmente positivos, sin embargo, no se observó correlación con reacciones adversas.
Anticuerpos contra PEG
Durante el programa de estudios clínicos, 37 pacientes tenían anticuerpos contra PEG antes de la administración de Esperoct. El test de anticuerpos contra PEG fue negativo en 20 de los 37 pacientes tras la administración del medicamento. En 17 pacientes se observó una respuesta temporal de bajo título de anticuerpos contra PEG. No se pudo establecer correlación con eventos adversos.
De acuerdo con los informes poscomercialización, también se observó la aparición de anticuerpos contra PEG durante el cambio a Esperoct. En algunos pacientes, los anticuerpos contra PEG podrían estar asociados con niveles de actividad de FVIII más bajos de lo esperado.
Pacientes pediátricos
No se han observado diferencias en el perfil de seguridad entre niños previamente tratados y adultos.
Se observó una reducción temporal de la recuperación incremental (IR) del nivel de factor VIII en algunos pacientes previamente no tratados, en ausencia de inhibidores de factor VIII (para más información, ver la sección «Instrucciones de uso»).
< u>Notificación de reacciones adversas y falta de eficacia del medicamento
La notificación de reacciones adversas tras la comercialización del medicamento es de gran importancia. Permite realizar un seguimiento continuo de la relación beneficio-riesgo del medicamento. Los profesionales médicos y farmacéuticos, así como los pacientes o sus representantes legales, deben notificar cualquier sospecha de reacción adversa o falta de eficacia del medicamento a través del Sistema Automatizado de Información de Farmacovigilancia en la siguiente dirección: https://aisf.dec.gov.ua.
Período de validez.
Frasco intacto (antes de la reconstitución):
36 meses si se conserva en nevera (2–8 °C).
Durante el período de validez, el medicamento puede conservarse:
- a temperatura ambiente (≤ 30 °C) desde el momento de fabricación hasta el inicio de la reconstitución durante un máximo de 12 meses
o
- por encima de la temperatura ambiente (de > 30 °C a 40 °C) desde el momento de fabricación hasta el inicio de la reconstitución durante un máximo de 3 meses.
Una vez que el medicamento ha sido almacenado fuera del refrigerador, no debe volver a colocarse en él.
Debe anotarse el inicio del almacenamiento fuera del refrigerador y la temperatura de almacenamiento en el espacio previsto en el envase exterior de cartón.
Después de la reconstitución:
La estabilidad química y física durante el uso ha sido demostrada:
- durante 24 horas si se conserva en nevera (2–8 °C) o
- durante 4 horas a ≤ 30 °C o
- durante 1 hora a temperaturas de > 30 °C hasta 40 °C, siempre que antes de la reconstitución el medicamento haya sido almacenado a temperaturas superiores a la temperatura ambiente (de > 30 °C a 40 °C) durante un período no superior a 3 meses.
Desde el punto de vista microbiológico, el medicamento debe usarse inmediatamente después de la reconstitución. Si el medicamento reconstituido no se utiliza inmediatamente, la responsabilidad sobre el tiempo y condiciones de almacenamiento durante su uso recae en el usuario; se recomienda no almacenar el medicamento más allá del tiempo indicado anteriormente, excepto cuando la reconstitución se realice bajo condiciones controladas y validadas que garanticen la esterilidad.
La solución reconstituida debe conservarse en el frasco.
Condiciones de almacenamiento.
Conservar en nevera (2–8 °C). No congelar. Conservar en el envase original para protegerlo de la luz.
Para obtener información sobre el almacenamiento a temperatura ambiente (≤ 30 °C) o a temperaturas hasta 40 °C, así como las condiciones de almacenamiento tras la reconstitución del medicamento, véase la sección «Período de validez». Mantener fuera del alcance de los niños.
Incompatibilidades.
Dado que no se han realizado estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos ni reconstituirse con otros soluciones inyectables distintas del diluyente suministrado en el envase del medicamento, cloruro de sodio.
El medicamento reconstituido no debe administrarse junto con otros medicamentos mediante el mismo sistema de perfusión ni a través del mismo recipiente que ya se esté utilizando para la administración de otros medicamentos.
Envase.
Cada envase de cartón del medicamento Esperoct contiene:
- 1 frasco de vidrio (vidrio tipo I) con polvo, cerrado con tapón de clorobutilo, tapa de aluminio y tapón plástico de desprendimiento;
- 1 dispositivo estéril de conexión para el frasco destinado a la reconstitución;
- 1 jeringa precargada de 4 ml con diluyente (solución de cloruro de sodio al 0,9 %) con sistema de cierre antirretorno (polipropileno), émbolo de goma (bromobutilo) y tapón de goma (bromobutilo);
- 1 émbolo (polipropileno).
Categoría de dispensación. Bajo receta médica.
Titular del registro/Producto.
A/T Novo Nordisk.
Domicilio del titular/productor y dirección del lugar de actividad.
Novo Allé,
Bagsværd, 2880,
Dinamarca.