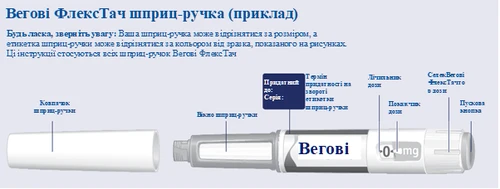
Vegovi flextech
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT Wegovy FlexTouch
Composition:
Active substance: semaglutide;
0.25 mg
each pre-filled pen contains 1 mg semaglutide* in 1.5 mL solution, 1 mL of solution contains 0.68 mg semaglutide*, 1 pre-filled pen contains 4 doses of 0.25 mg each;
0.5 mg
each pre-filled pen contains 2 mg semaglutide* in 1.5 mL solution, 1 mL of solution contains 1.34 mg semaglutide*, 1 pre-filled pen contains 4 doses of 0.5 mg each;
1 mg
each pre-filled pen contains 4 mg semaglutide* in 3 mL solution, 1 mL of solution contains 1.34 mg semaglutide*, 1 pre-filled pen contains 4 doses of 1 mg each;
1.7 mg
each pre-filled pen contains 6.8 mg semaglutide* in 3 mL solution, 1 mL of solution contains 2.27 mg semaglutide*, 1 pre-filled pen contains 4 doses of 1.7 mg each;
2.4 mg
each pre-filled pen contains 9.6 mg semaglutide* in 3 mL solution, 1 mL of solution contains 3.2 mg semaglutide*, 1 pre-filled pen contains 4 doses of 2.4 mg each;
Excipients: disodium phosphate dihydrate; propylene glycol; phenol; hydrochloric acid (for pH adjustment); sodium hydroxide (for pH adjustment); water for injections.
* A human glucagon-like peptide-1 (GLP-1) analogue produced by recombinant DNA technology in Saccharomyces cerevisiae.
Pharmaceutical form. Solution for injection.
Main physicochemical properties: clear, colorless, isotonic solution; pH = 7.4.
Pharmacotherapeutic group. Blood glucose lowering agents, excluding insulin. Glucagon-like peptide-1 (GLP-1) analogues. ATC code A10BJ06.
Pharmacological Properties.
Pharmacodynamics.
Mechanism of action
Semaglutide is a GLP-1 analogue with 94% homology to human GLP-1. Semaglutide acts as a GLP-1 receptor agonist, selectively binding to and activating GLP-1 receptors, which are the target for native GLP-1.
GLP-1 is a physiological regulator of appetite and caloric intake, and GLP-1 receptors are present in several brain regions involved in appetite regulation.
Animal studies show that semaglutide acts in the brain via the GLP-1 receptor. Semaglutide has a direct effect on brain areas involved in homeostatic regulation of food intake in the hypothalamus and brainstem. Semaglutide may influence the hedonic reward system through direct and indirect effects on brain regions including the septum, thalamus, and amygdala.
Clinical studies show that semaglutide reduces energy intake, increases feelings of satiety, fullness, and control over eating, reduces hunger, as well as the frequency and intensity of food cravings. In addition, semaglutide reduces the craving for high-fat foods.
Semaglutide regulates both homeostatic and hedonic inputs with executive function to control calorie intake, appetite, reward, and food choice.
Furthermore, clinical studies have shown that semaglutide reduces blood glucose levels by glucose-dependent stimulation of insulin secretion and suppression of glucagon secretion under conditions of elevated blood glucose concentration. The mechanism of blood glucose reduction is also accompanied by a slight delay in gastric emptying during the early postprandial phase. During hypoglycemia, semaglutide reduces insulin secretion and does not interfere with glucagon secretion.
GLP-1 receptor expression also occurs in the heart, vascular system, immune system, and kidneys.
In clinical studies, semaglutide had a positive effect on plasma lipid levels, reduced systolic blood pressure, and decreased inflammation. In addition, animal studies show that semaglutide inhibited the development of atherosclerosis and exhibited anti-inflammatory effects on the cardiovascular system.
Pharmacodynamic effects
Appetite, energy intake, and food choice
Semaglutide reduces appetite by enhancing feelings of satiation and fullness, while simultaneously reducing hunger and potential food intake. In a phase 1 study, energy intake during ad libitum meals was 35% lower with semaglutide compared to placebo after 20 weeks of treatment. This was supported by improved control over eating, reduced food cravings, and relatively lower cravings for high-fat foods. Food cravings were further assessed in the STEP 5 trial using the Control of Eating Questionnaire (CoEQ). At week 104, the calculated treatment difference favored semaglutide for both control of food cravings and cravings for salty foods, while no clear effect on cravings for sweet foods was observed.
Fasting and postprandial lipids
Compared to placebo, semaglutide at a dose of 1 mg reduced fasting triglyceride and very-low-density lipoprotein (VLDL) cholesterol concentrations by 12% and 21%, respectively. Postprandial levels of triglycerides and VLDL in response to a high-fat meal were reduced by more than 40%.
Clinical efficacy and safety
The efficacy and safety of semaglutide for weight management, as an adjunct to a reduced-calorie diet and increased physical activity, were evaluated in four double-blind, randomized, placebo-controlled phase 3a trials of 68 weeks duration (STEP 1–4). A total of 4,684 patients (2,652 randomized to semaglutide treatment) were included in these trials. Additionally, the two-year efficacy and safety of semaglutide compared to placebo were evaluated in a double-blind, randomized, placebo-controlled phase 3b trial (STEP 5), which included 304 patients (152 patients receiving semaglutide treatment).
Treatment with semaglutide demonstrated superior, clinically meaningful, and sustained weight reduction compared to placebo in patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and at least one weight-related comorbidity. Furthermore, a greater proportion of patients achieved ≥ 5%, ≥ 10%, ≥ 15%, and ≥ 20% weight loss with semaglutide compared to placebo. Weight reduction occurred independently of gastrointestinal symptoms such as nausea, vomiting, or diarrhea.
Treatment with semaglutide also showed statistically significant improvements in waist circumference, systolic blood pressure, and level of physical activity compared to placebo.
Efficacy was demonstrated regardless of patient age, sex, race, ethnicity, baseline body weight, BMI, presence of type 2 diabetes, or degree of renal impairment. Variations in efficacy were observed across all patient subgroups. Relatively greater weight loss was observed in women and in patients without type 2 diabetes, as well as in patients with lower baseline body weight compared to higher.
STEP 1 trial: Weight management
In a double-blind, 68-week clinical trial, 1,961 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and at least one weight-related comorbidity were randomized to receive either semaglutide or placebo. All patients followed a reduced-calorie diet and increased physical activity throughout the study.
Weight reduction occurred early and continued throughout the study. At the end of treatment (week 68), weight reduction was superior and clinically significant compared to placebo (see Table 1 and Figure 1). Additionally, a greater proportion of patients achieved ≥ 5%, ≥ 10%, ≥ 15%, and ≥ 20% weight loss with semaglutide compared to placebo (see Table 1). Among patients with baseline prediabetes, a greater proportion achieved normoglycemic status at the end of treatment with semaglutide compared to placebo (84.1% vs. 47.8%).
Table 1. STEP 1 trial: Results at week 68
| Parameter |
Wegovy FlexTouch |
Placebo |
| Number of patients (N) |
1306 |
655 |
| Body weight (kg) |
||
| Baseline level (kg) |
105.4 |
105.2 |
| Change (%) from baseline1,2 |
-14.9 |
-2.4 |
| Difference (%) vs placebo1 [95% CI] |
-12.4 [-13.4; -11.5]* |
- |
| Change (kg) from baseline |
-15.3 |
-2.6 |
| Difference (kg) vs placebo1 [95% CI] |
-12.7 [-13.7; -11.7] |
- |
| Patients (%) achieving body weight loss ≥ 5%3 |
83.5* |
31.1 |
| Patients (%) achieving body weight loss ≥ 10%3 |
66.1* |
12.0 |
| Patients (%) achieving body weight loss ≥ 15%3 |
47.9* |
4.8 |
| Waist circumference (cm) |
||
| Baseline level |
114.6 |
114.8 |
| Change from baseline1 |
-13.5 |
-4.1 |
| Difference vs placebo1 [95% CI] |
-9.4 [-10.3; -8.5]* |
- |
| Systolic blood pressure (mmHg) |
||
| Baseline level |
126 |
127 |
| Change from baseline1 |
-6.2 |
-1.1 |
| Difference vs placebo1 [95% CI] |
-5.1 [-6.3; -3.9]* |
- |
* p < 0.0001 (unadjusted two-sided) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the trial, treatment was permanently discontinued in 17.1% and 22.4% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -16.9% and -2.4% for semaglutide 2.4 mg and placebo, respectively.
3 Estimated using a binary regression model based on the same imputation procedure as in the primary analysis.
HTMLIMG0END
Observed values for patients completing each scheduled visit and multiple imputation (MI) estimates accounting for data from patients who discontinued.
Fig. 1. STEP 1: Mean change in body weight (%) from baseline to week 68
After the 68-week study, a 52-week treatment-free period was conducted, involving 327 patients who completed the main study period on maintenance dose of semaglutide or placebo. During the treatment-free period from week 68 to week 120, mean body weight increased in both treatment groups. However, in patients who had received semaglutide during the main study period, body weight remained 5.6% lower than baseline compared to 0.1% in the placebo group.
Study STEP 2. Weight control in patients with type 2 diabetes
In a double-blind, 68-week clinical trial, 1210 patients with overweight or obesity (BMI ≥ 27 kg/m²) and type 2 diabetes were randomized to receive once-weekly semaglutide 2.4 mg, semaglutide 1 mg, or placebo. Patients enrolled in the study had inadequately controlled type 2 diabetes (HbA1c 7–10%) and were either on diet and exercise alone or on 1–3 oral antidiabetic agents. All patients followed a reduced-calorie diet and increased physical activity throughout the study.
Treatment with semaglutide for 68 weeks resulted in significant and clinically meaningful reductions in body weight and HbA1c compared to placebo (see Table 2 and Figure 2).
Table 2. Study STEP 2: Results at Week 68
| Parameter |
Wegovy FlexTouch |
Placebo |
| Number of patients (N) |
404 |
403 |
| Body weight (kg) |
||
| Baseline (kg) |
99.9 |
100.5 |
| Change (%) from baseline1,2 |
-9.6 |
-3.4 |
| Difference (%) vs placebo1 [95% CI] |
-6.2 [-7.3; -5.2]* |
- |
| Change (kg) from baseline |
-9.7 |
-3.5 |
| Difference (kg) vs placebo1 [95% CI] |
-6.1 [-7.2; -5.0] |
- |
| Patients (%) achieving body weight loss ≥ 5%3 |
67.4* |
30.2 |
| Patients (%) achieving body weight loss ≥ 10%3 |
44.5* |
10.2 |
| Patients (%) achieving body weight loss ≥ 15%3 |
25.0* |
4.3 |
| Waist circumference (cm) |
||
| Baseline |
114.5 |
115.5 |
| Change from baseline1 |
-9.4 |
-4.5 |
| Difference vs placebo1 [95% CI] |
-4.9 [-6.0; -3.8]* |
- |
| Systolic blood pressure (mmHg) |
||
| Baseline |
130 |
130 |
| Change from baseline1 |
-3.9 |
-0.5 |
| Difference vs placebo1 [95% CI] |
-3.4 [-5.6; -1.3]** |
- |
| HbA1c (mmol/mol (%)) |
||
| Baseline |
65.3 (8.1) |
65.3 (8.1) |
| Change from baseline1 |
-17.5 (-1.6) |
-4.1 (-0.4) |
| Difference vs placebo1 [95% CI] |
-13.5 [-15.5; -11.4] (-1.2 [-1.4; -1.1])* |
- - |
* p < 0.0001 (unadjusted two-sided) in favor; ** p < 0.05 (unadjusted two-sided) in favor.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the trial, treatment was permanently discontinued in 11.6% and 13.9% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -10.6% and -3.1% for semaglutide 2.4 mg and placebo, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
**
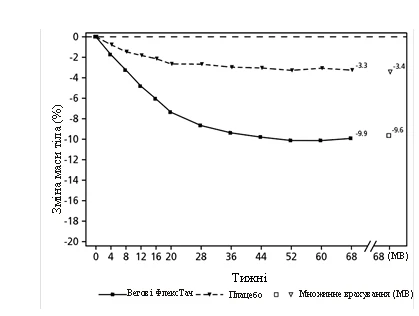
**
Observed values for patients completing each scheduled visit and multiple imputation (MI) estimates for data from patients who discontinued.
Fig. 2. STEP 2: Mean change in body weight (%) from baseline to week 68
Study STEP 3. Weight Management with Intensive Behavioral Therapy
In a double-blind, 68-week clinical trial, 611 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and at least one weight-related comorbidity were randomized to receive either semaglutide or placebo. All patients received intensive behavioral therapy (IPT), consisting of a highly energy-restricted diet, increased physical activity, and behavioral counseling throughout the study.
Treatment with semaglutide and IPT over 68 weeks resulted in significant and clinically meaningful weight reduction compared to placebo (see Table 3).
Table 3. Study STEP 3: Results at Week 68
| Parameter |
Wegovy FlexTouch |
Placebo |
| Number of patients (N) |
407 |
204 |
| Body weight (kg) |
||
| Baseline (kg) |
106.9 |
103.7 |
| Change (%) from baseline1,2 |
-16.0 |
-5.7 |
| Difference (%) vs placebo1 [95% CI] |
-10.3 [-12.0; -8.6]* |
- |
| Change (kg) from baseline |
-16.8 |
-6.2 |
| Difference (kg) vs placebo1 [95% CI] |
-10.6 [-12.5; -8.8] |
- |
| Patients (%) achieving body weight loss ≥ 5%3 |
84.8* |
47.8 |
| Patients (%) achieving body weight loss ≥ 10%3 |
73.0* |
27.1 |
| Patients (%) achieving body weight loss ≥ 15%3 |
53.5* |
13.2 |
| Waist circumference (cm) |
||
| Baseline |
113.6 |
111.8 |
| Change from baseline1 |
-14.6 |
-6.3 |
| Difference vs placebo1 [95% CI] |
-8.3 [-10.1; -6.6]* |
- |
| Systolic blood pressure (mm Hg) |
||
| Baseline |
124 |
124 |
| Change from baseline1 |
-5.6 |
-1.6 |
| Difference vs placebo1 [95% CI] |
-3.9 [-6.4; -1.5]* |
- |
*p < 0.0001 (unadjusted two-sided) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 16.7% and 18.6% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -17.6% and -5.0% for semaglutide 2.4 mg and placebo, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
STEP 4 Study. Maintenance of Body Weight Control
In a double-blind, 68-week clinical trial, 902 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and at least one weight-related comorbidity were enrolled. All patients followed a reduced-calorie diet and increased physical activity throughout the study. From week 0 to week 20 (run-in period), all patients received semaglutide. At week 20 (baseline), patients who had reached the maintenance dose of 2.4 mg were randomized to continue treatment or switch to placebo. At week 0 (start of run-in period), patients had a mean body weight of 107.2 kg and a mean BMI of 38.4 kg/m².
Patients who reached the maintenance dose of 2.4 mg at week 20 (baseline) and continued semaglutide treatment for 48 weeks (weeks 20–68) continued to lose body weight and achieved greater and clinically meaningful weight reduction compared to those who switched to placebo (see Table 4 and Figure 3). Body weight steadily increased from week 20 to week 68 in patients who switched to placebo at week 20 (baseline). However, the observed mean body weight at week 68 was still lower than at the start of the run-in period (week 0) (see Figure 3). Patients who received semaglutide from week 0 (run-in) to week 68 (end of treatment) achieved a mean change in body weight of -17.4%, with 87.8% achieving weight loss ≥ 5%, 78.0% achieving ≥ 10%, 62.2% achieving ≥ 15%, and 38.6% achieving ≥ 20%.
Table 4. STEP 4 Study: Results from Week 20 to Week 68
| Parameter |
Wegovy FlexTouch |
Placebo |
| Number of patients (N) |
535 |
268 |
| Body weight (kg) |
||
| Baseline level1 (kg) |
96.5 |
95.4 |
| Change (%) from baseline1,2,3 |
-7.9 |
6.9 |
| Difference (%) vs placebo2 [95% CI] |
-14.8 [-16.0; -13.5]* |
- |
| Change (kg) from baseline |
-7.1 |
6.1 |
| Difference (kg) vs placebo2 [95% CI] |
-13.2 [-14.3; -12.0] |
- |
| Waist circumference (cm) |
||
| Baseline level |
105.5 |
104.7 |
| Change from baseline1 |
-6.4 |
3.3 |
| Difference vs placebo2 [95% CI] |
-9.7 [-10.9; -8.5]* |
- |
| Systolic blood pressure (mmHg) |
||
| Baseline level |
121 |
121 |
| Change from baseline1,2 |
0.5 |
4.4 |
| Difference vs placebo2 [95% CI] |
-3.9 [-5.8; -2.0]* |
- |
* p < 0.0001 (unadjusted two-sided) for superiority.
1 Baseline = week 20.
2 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
3 During the trial, treatment was permanently discontinued in 5.8% and 11.6% of patients randomized to semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from randomization to week 68 in body weight based on a mixed model for repeated measures, including all observations up to first discontinuation, were -8.1% and -6.5% for semaglutide 2.4 mg and placebo, respectively.
**
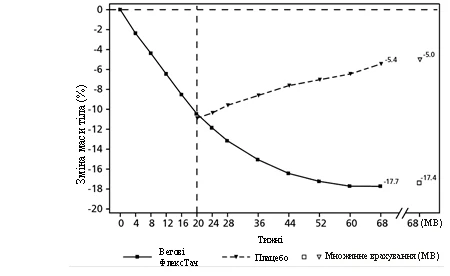
**
Observed values for patients completing each scheduled visit and multiple imputation (MI) estimates accounting for data from patients who discontinued.
Fig. 3. STEP 4: Mean change in body weight (%) from week 0 to week 68
Study STEP 5. 2-year data
In a double-blind, 104-week clinical trial, 304 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and at least one weight-related comorbidity were randomized to receive semaglutide or placebo. All patients followed a reduced-calorie diet and increased physical activity throughout the study. At baseline, patients had a mean BMI of 38.5 kg/m² and a mean body weight of 106.0 kg.
Treatment with semaglutide for 104 weeks resulted in significant and clinically meaningful weight reduction compared to placebo. Mean body weight decreased from baseline to week 68 with semaglutide, after which a plateau was reached. With placebo, mean body weight decreased to a lesser extent and plateaued at approximately 20 weeks of treatment (see Table 5 and Figure 4). Patients receiving semaglutide achieved a mean change in body weight of -15.2%, with 74.7% of these patients losing ≥ 5% body weight, 59.2% losing ≥ 10%, and 49.7% losing ≥ 15%. 80% and 37% of patients with prediabetes at baseline achieved normal glycemic status at the end of treatment with semaglutide and placebo, respectively.
Table 5. Study STEP 5: Results at Week 104
| Parameter |
Wegovy FlexTouch |
Placebo |
| Number of patients (N) |
152 |
152 |
| Body weight (kg) |
||
| Baseline level (kg) |
105.6 |
106.5 |
| Change (%) from baseline1,2 |
-15.2 |
-2.6 |
| Difference (%) vs placebo1 [95% CI] |
-12.6 [-15.3; -9.8]* |
- |
| Change (kg) from baseline |
-16.1 |
-3.2 |
| Difference (kg) vs placebo1 [95% CI] |
-12.9 [-16.1; -9.8] |
- |
| Patients (%) achieving body weight loss ≥ 5%3 |
74.7* |
37.3 |
| Patients (%) achieving body weight loss ≥ 10%3 |
59.2* |
16.8 |
| Patients (%) achieving body weight loss ≥ 15%3 |
49.7* |
9.2 |
| Waist circumference (cm) |
||
| Baseline level |
115.8 |
115.7 |
| Change from baseline1 |
-14.4 |
-5.2 |
| Difference vs placebo1 [95% CI] |
-9.2 [-12.2; -6.2]* |
- |
| Systolic blood pressure (mmHg) |
||
| Baseline level |
126 |
125 |
| Change from baseline1 |
-5.7 |
-1.6 |
| Difference vs placebo1 [95% CI] |
-4.2 [-7.3; -1.0]* |
- |
* p < 0.0001 (unadjusted two-sided) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 13.2% and 27.0% of patients randomized to receive semaglutide and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -16.7% and -0.6% for semaglutide and placebo, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
HTMLIMG3END
Observed values for patients completing each scheduled visit and multiple imputation (MI) estimates accounting for data from patients who discontinued.
Fig. 4. STEP 5: Mean change in body weight (%) from week 0 to week 104
Study STEP 8. Comparison of semaglutide with liraglutide
In a randomized, open-label, pairwise placebo-controlled trial of 68 weeks' duration, 338 patients with obesity (BMI ≥ 30 kg/m²) or overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) and with at least one weight-related comorbidity were randomized to receive once-weekly semaglutide, once-daily liraglutide 3 mg, or placebo. Once-weekly semaglutide and liraglutide 3 mg were administered openly, but each active treatment group was double-blind to placebo administered at the same dosing frequency. At baseline, patients had a mean BMI of 37.5 kg/m² and a mean body weight of 104.5 kg.
Treatment with once-weekly semaglutide for 68 weeks resulted in significantly greater and clinically meaningful weight reduction compared to liraglutide. Mean body weight decreased from baseline to week 68 with semaglutide treatment. With liraglutide treatment, mean body weight decreased to a lesser extent (see Table 6). 37.4% of patients receiving semaglutide lost ≥ 20% of body weight, compared to 7.0% of patients receiving liraglutide. Table 6 presents the results for confirmatory endpoints of ≥ 10%, ≥ 15%, and ≥ 20% body weight loss.
Table 6. Study STEP 8: Results at week 68 comparing semaglutide and liraglutide
| Parameter |
Wegovy FlexTouch |
Liraglutide 3 mg |
| Number of patients (N) |
126 |
127 |
| Body weight (kg) |
||
| Baseline (kg) |
102.5 |
103.7 |
| Change (%) from baseline1,2 |
-15.8 |
-6.4 |
| Difference (%) vs liraglutide1 [95% CI] |
-9.4 [-12.0; -6.8]* |
- |
| Change (kg) from baseline |
-15.3 |
-6.8 |
| Difference (kg) vs liraglutide1 [95% CI] |
-8.5 [-11.2; -5.7] |
- |
| Patients (%) achieving body weight loss ≥ 10%3 |
69.4* |
27.2 |
| Patients (%) achieving body weight loss ≥ 15%3 |
54.0* |
13.4 |
| Patients (%) achieving body weight loss ≥ 20%3 |
37.4* |
7.0 |
* p < 0.0001 (unadjusted two-sided) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the trial, treatment was permanently discontinued in 13.5% and 27.6% of patients randomized to semaglutide and liraglutide, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from randomization to week 68 in body weight based on a mixed model for repeated measurements, including all observations up to first discontinuation, were -16.7% and -6.7% for semaglutide and liraglutide, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
Effect on Body Composition
In a sub-study of STEP 1 (N = 140), body composition was measured using dual-energy X-ray absorptiometry (DEXA). DEXA assessment results showed that treatment with semaglutide was associated with a greater reduction in fat mass than in lean body mass, leading to improved body composition compared to placebo at week 68. Furthermore, the reduction in total fat mass was accompanied by a reduction in visceral fat. These results indicate that the majority of overall body weight loss was related to a reduction in adipose tissue, including visceral fat.
Improvement in Physical Functioning
Semaglutide demonstrated a modest improvement in physical functioning outcomes. Physical functioning was assessed using both the general health-related quality of life questionnaire, Short Form-36v2 Health Survey, Acute Version (SF-36), and the Impact of Weight on Quality of Life Clinical Trials Version (IWQOL-Lite-CT).
Cardiovascular Assessment
In the SUSTAIN 6 trial, 3297 patients with inadequately controlled type 2 diabetes and high cardiovascular risk were randomized to receive subcutaneous semaglutide 0.5 mg or 1 mg once weekly or placebo added to standard therapy. The treatment duration was 104 weeks. The mean age of patients was 65 years, and the mean BMI was 33 kg/m².
The primary endpoint was time from randomization to the first occurrence of a major adverse cardiovascular event (MACE): cardiovascular death, non-fatal myocardial infarction, or non-fatal stroke. The total number of MACE events was 254, including 108 (6.6%) with semaglutide and 146 (8.9%) with placebo.
Cardiovascular safety of semaglutide 0.5 or 1 mg was confirmed, as the hazard ratio (HR) for semaglutide versus placebo was 0.74 [0.58, 0.95] [95% CI], driven by a reduction in non-fatal myocardial infarction and non-fatal stroke, with no difference in the rate of cardiovascular death (see Figure 5).
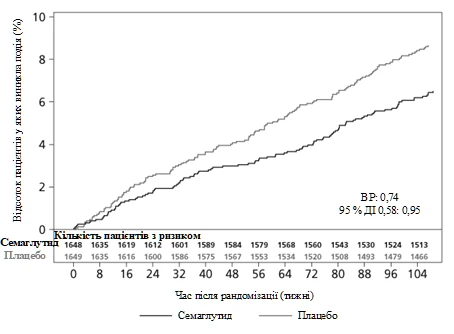
Figure 5. Kaplan-Meier plot of time to first occurrence of the composite endpoint: cardiovascular death, non-fatal myocardial infarction, and non-fatal stroke (clinical trial SUSTAIN 6)
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Wegovy FlexTouch in one or more subsets of the paediatric population for weight control (see section "Posology and method of administration" for information on use in children).
STEPTEENS study: Weight control in patients aged 12–18 years
In a 68-week double-blind trial, 201 pubertal children aged 12 to 18 years with obesity or overweight and at least one weight-related comorbidity were randomized 2:1 to receive semaglutide or placebo. All patients followed a reduced-calorie diet and increased physical activity throughout the study.
At the end of treatment (week 68), improvement in BMI with semaglutide was greater and clinically meaningful compared to placebo (see Table 6 and Figure 6). Additionally, a higher proportion of patients achieved ≥ 5%, ≥ 10%, and ≥ 15% body weight loss with semaglutide compared to placebo (see Table 6).
Table 6. STEPTEENS: Outcomes at Week 68
| Parameter |
Wegovy FlexTouch |
Placebo |
| Number of patients (N) |
134 |
67 |
| BMI |
||
| Baseline (BMI) |
37.7 |
35.7 |
| Change (%) from baseline1,2 |
-16.1 |
0.6 |
| Difference (%) vs placebo1 [95% CI] |
-16.7 [-20.3; -13.2] |
- |
| Baseline (BMI SDS (standard deviation score)) |
3.4 |
3.1 |
| Change from baseline BMI SDS1 |
-1.1 |
-0.1 |
| Difference vs placebo1 [95% CI] |
-1.0 [-1.3; -0.8] |
- |
| Body Weight |
||
| Baseline (kg) |
109.9 |
102.6 |
| Change (%) from baseline1 |
-14.7 |
2.8 |
| Difference (%) vs placebo1 [95% CI] |
-17.4 [-21.1; -13.8] |
- |
| Change (kg) from baseline1 |
-15.3 |
2.4 |
| Difference (kg) vs placebo1 [95% CI] |
-17.7 [-21.8; -13.7] |
- |
| Patients (%) achieving body weight loss ≥ 5%3 |
72.5* |
17.7 |
| Number of patients (N) |
134 |
67 |
| Patients (%) achieving body weight loss ≥ 10%3 |
61.8 |
8.1 |
| Patients (%) achieving body weight loss ≥ 15%3 |
53.4 |
4.8 |
| Waist circumference (cm) |
||
| Baseline |
111.9 |
107.3 |
| Change from baseline1 |
-12.7 |
-0.6 |
| Difference vs placebo1 [95% CI] |
-12.1 [-15.6; -8.7]* |
- |
| Systolic blood pressure (mmHg) |
||
| Baseline |
120 |
120 |
| Change from baseline1 |
-2.7 |
-0.8 |
| Difference vs placebo1 [95% CI] |
-1.9 [-5.0; -1.1]* |
- |
*p < 0.0001 (unadjusted two-sided) for superiority.
1 Assessed using an ANCOVA model with multiple imputation based on all data, regardless of discontinuation of randomized treatment or initiation of other anti-obesity medications or bariatric surgery.
2 During the study, treatment was permanently discontinued in 10.4% and 10.4% of patients randomized to receive semaglutide 2.4 mg and placebo, respectively. Assuming all randomized patients continued treatment and did not receive additional anti-obesity therapy, estimated changes from baseline to week 68 in body weight based on a mixed model for repeated measures, including all observations up to first discontinuation, were -17.9% and -0.6% for semaglutide 2.4 mg and placebo, respectively.
3 Assessed using a binary regression model based on the same imputation procedure as in the primary analysis.
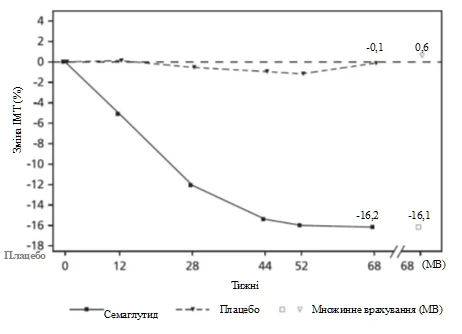
Observed values for patients completing each scheduled visit and multiple imputation (MI) estimates for data from patients who discontinued.
Fig. 6. STEP TEENS: Mean change in BMI (%) from baseline to week 68
Pharmacokinetics.
Compared to native GLP-1, semaglutide has a prolonged elimination half-life of approximately 1 week, allowing for once-weekly subcutaneous administration. The primary mechanism underlying this prolonged action is albumin binding, which reduces renal clearance and protects against metabolic degradation. Additionally, semaglutide is stabilized against degradation by the enzyme DPP-4.
Absorption
The mean steady-state concentration of semaglutide following subcutaneous administration of its maintenance dose was approximately 75 nmol/L in patients with overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) or obesity (BMI ≥ 30 kg/m²), based on phase 3 trial data, where 90% of patients had concentrations ranging from 51 nmol/L to 110 nmol/L. With doses ranging from 0.25 mg to 2.4 mg administered once weekly, steady-state exposure to semaglutide increased in a dose-dependent manner. Steady-state exposure was stable over time, as determined up to week 68. Similar exposure was achieved following subcutaneous administration of semaglutide in the abdominal wall, thigh, or upper arm. The absolute bioavailability of semaglutide following subcutaneous administration was 89%.
Distribution
The mean volume of distribution of semaglutide after subcutaneous administration in patients with overweight or obesity was approximately 12.4 L. Semaglutide is highly bound to plasma albumin (> 99%).
Metabolism/Biotransformation
Prior to excretion, semaglutide is actively metabolized via proteolytic cleavage of the peptide backbone and subsequent beta-oxidation of the fatty acid side chain. Neutral endopeptidase (NEP) has been identified as one of the active metabolic enzymes.
Elimination
Material related to semaglutide is primarily excreted in urine and feces. Approximately 3% of the administered dose was excreted in urine as unchanged semaglutide. The clearance of semaglutide in patients with overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) or obesity (BMI ≥ 30 kg/m²) was approximately 0.05 L/h. With its half-life of approximately 1 week, semaglutide will remain in circulation for about 7 weeks after the last 2.4 mg dose.
Special patient populations
Geriatric patients
Patient age does not affect the pharmacokinetics of semaglutide, based on phase 3 trial data involving patients aged 18–86 years.
Sex, race, and ethnicity
Patient sex, race (Caucasian, African, or Asian), and ethnicity (Hispanic or Latino, non-Hispanic or non-Latino) do not affect the pharmacokinetics of semaglutide, based on phase 3a trial data.
Body weight
Patient body weight affects semaglutide exposure. Higher body weight leads to lower drug exposure; a 20% difference in body weight between patients results in nearly an 18% difference in exposure. The semaglutide dose of 2.4 mg once weekly provides adequate systemic exposure across a body weight range of 54.4–245.6 kg, as evaluated for exposure-response relationships in clinical trials.
Renal impairment
Renal impairment does not have a clinically significant effect on the pharmacokinetics of semaglutide. This was demonstrated following a single 0.5 mg dose of semaglutide in patients with varying degrees of renal impairment (mild, moderate, severe, and dialysis-dependent) compared to patients with normal renal function. This was also confirmed by phase 3a clinical trial data in patients with overweight (BMI ≥ 27 kg/m² to < 30 kg/m²) or obesity (BMI ≥ 30 kg/m²) and mild to moderate renal impairment.
Hepatic impairment
Hepatic impairment does not affect semaglutide exposure. The pharmacokinetics of semaglutide were evaluated in patients with varying degrees of hepatic impairment (mild, moderate, severe) compared to patients with normal liver function in a single-dose 0.5 mg semaglutide study.
Prediabetes and diabetes
Prediabetes and diabetes do not have any clinically significant effect on semaglutide exposure, based on phase 3 trial data.
Immunogenicity
Antibody development against semaglutide during treatment occurred infrequently (see section "Adverse reactions"), and the immune response did not appear to affect the pharmacokinetics of semaglutide.
Children
The pharmacokinetic properties of semaglutide were evaluated in a clinical trial involving adolescent patients (aged 12 to < 18 years, n=124, body weight 61.6–211.9 kg) with obesity or overweight and at least one comorbidity related to body weight. Semaglutide exposure in children was similar to that observed in adults with obesity or overweight.
The safety and efficacy of semaglutide in children under 12 years of age have not been studied.
Preclinical safety data
Preclinical data based on studies of pharmacological safety, repeated-dose toxicity, and genotoxicity revealed no risk for humans.
Non-lethal tumors originating from C-cells of the thyroid gland observed in rodents are effects specific to the class of GLP-1 receptor agonists. In a 2-year carcinogenicity study in rats and mice, semaglutide caused the development of thyroid C-cell tumors at clinically relevant exposure levels. No other treatment-related tumors were observed. The development of C-cell tumors in rodents is due to a non-genotoxic, GLP-1 receptor-mediated mechanism to which rodents are partially sensitive. The relevance of this mechanism to humans is considered low but cannot be entirely excluded.
In fertility studies in rats, semaglutide did not affect mating performance or fertility in males. In female rats, prolongation of the estrous cycle and a slight reduction in corpora lutea (ovulation) were observed at doses associated with body weight loss.
In embryo-fetal development studies in rats, semaglutide caused embryotoxic effects at exposures lower than clinically relevant levels. Semaglutide caused significant body weight loss in females and reduced embryonic survival and growth. Fetal skeletal and visceral malformations were observed, including changes in long bones, ribs, spine, tail bones, blood vessels, and brain ventricles. Mechanistic evaluation indicated that the embryotoxic effect involves GLP-1 receptor-mediated impairment of nutrient supply to the embryo via the yolk sac in rats. Due to anatomical and functional differences in the yolk sac across species and the lack of GLP-1 receptor expression in the yolk sac of non-human primates, this mechanism is considered unlikely in humans. However, a direct effect of semaglutide on the fetus cannot be ruled out.
In developmental toxicity studies in rabbits and cynomolgus monkeys at clinically relevant exposure levels, increased rates of pregnancy loss and some increase in fetal abnormalities were observed. These findings coincided with significant body weight loss in females, reaching up to 16%. It is unclear whether these effects are related to reduced food intake due to the direct action of GLP-1.
Postnatal growth and development were assessed in cynomolgus monkeys. Offspring were slightly smaller at birth, but their body weight normalized during the lactation period.
In young rats, semaglutide caused delayed sexual maturation in both males and females. This delay did not affect fertility or reproductive capacity in either sex, nor the ability of females to maintain pregnancy.
Clinical characteristics.
Indications
Adults
The medicinal product Wegovy FlexTouch is indicated for weight control as an adjunct to a reduced-calorie diet and increased physical activity in adult patients with an initial body mass index (BMI) of
- greater than 30 kg/m² (obesity), or
- from 27 to 30 kg/m² (overweight) in the presence of at least one weight-related comorbid condition, such as dysglycemia (prediabetes or type 2 diabetes), hypertension, dyslipidemia, obstructive sleep apnea, or cardiovascular disease.
Children ≥ 12 years
The medicinal product Wegovy FlexTouch is indicated for weight control as an adjunct to a reduced-calorie diet and increased physical activity in children aged 12 years and older with
- obesity* and
- body weight above 60 kg.
Wegovy FlexTouch treatment should be discontinued and reassessed if BMI has not decreased by at least 5% after 12 weeks of treatment with 2.4 mg or the maximum tolerated dose.
* Obesity (BMI ≥ 95th percentile), as defined by sex- and age-specific BMI growth charts (CDC.gov) (see Table 7).
Table 7. BMI thresholds for obesity (≥ 95th percentile) by sex and age for children aged 12 years and older (CDC criteria)
| Age (years) |
BMI (kg/m²) at the 95th percentile |
|
| Male |
Female |
|
| 12 |
24.2 |
25.2 |
| 12.5 |
24.7 |
25.7 |
| 13 |
25.1 |
26.3 |
| 13.5 |
25.6 |
26.8 |
| 14 |
26.0 |
27.2 |
| 14.5 |
26.4 |
27.7 |
| 15 |
26.8 |
28.1 |
| 15.5 |
27.2 |
28.5 |
| 16 |
27.5 |
28.9 |
| 16.5 |
27.9 |
29.3 |
| 17 |
28.2 |
29.6 |
| 17.5 |
28.6 |
30.0 |
Contraindications.
Hypersensitivity to the active substance or to any of the excipients of the medicinal product (see section "Composition").
Interaction with other medicinal products and other forms of interaction.
Semaglutide delays gastric emptying and has the potential to affect the absorption rate of concomitantly administered oral medicinal products. No clinically significant effect on gastric emptying rate has been observed after administration of semaglutide 2.4 mg, likely due to a tachyphylaxis effect. Semaglutide should be used with caution in patients receiving oral medicinal products that require rapid absorption in the gastrointestinal tract.
Paracetamol
Based on the pharmacokinetic assessment of paracetamol during a standardized meal test, semaglutide delays the rate of gastric emptying. After concomitant administration of paracetamol with semaglutide 1 mg, AUC0–60min and Cmax of paracetamol were reduced by 27% and 23%, respectively. Overall exposure to paracetamol (AUC0–5h) was unchanged. No clinically significant effect of semaglutide on paracetamol was observed. Dose adjustment is not required when paracetamol is administered concomitantly with semaglutide.
Oral contraceptives
A reduction in the efficacy of oral contraceptives by semaglutide is not expected. When administered concomitantly with a combined oral contraceptive medicinal product (ethinylestradiol 0.03 mg/levonorgestrel 0.15 mg), semaglutide did not clinically significantly affect the overall exposure of ethinylestradiol and levonorgestrel. Exposure to ethinylestradiol was unaffected; exposure to levonorgestrel increased by 20% at steady state. Cmax of either component was unchanged.
Atorvastatin
Semaglutide did not alter the overall exposure of atorvastatin following a single dose of atorvastatin (40 mg). Cmax of atorvastatin was reduced by 38%. This was considered clinically insignificant.
Digoxin
Semaglutide did not alter the overall exposure or Cmax of digoxin following a single dose of digoxin (0.5 mg).
Metformin
Semaglutide did not alter the overall exposure or Cmax of metformin following administration of metformin 500 mg twice daily for 3.5 days.
Warfarin
Semaglutide did not alter the overall exposure or Cmax of R- and S-warfarin following a single dose of warfarin (25 mg), and the pharmacodynamic effects of warfarin, measured by the international normalized ratio (INR), were not clinically significantly changed. However, frequent monitoring of INR is recommended at the beginning of semaglutide treatment in patients taking warfarin or other coumarin derivatives.
Children
Interaction studies have only been conducted in adult patients.
Special precautions for use.
Traceability
In order to improve the traceability of biological medicinal products, the name and batch number of the administered product should be clearly recorded on the packaging.
Dehydration
Use of GLP-1 receptor agonists may be associated with gastrointestinal adverse reactions, which may lead to dehydration; in rare cases, this may result in worsening renal function. Patients should be advised of the potential risk of dehydration due to gastrointestinal adverse reactions and should take measures to avoid dehydration.
Acute pancreatitis
Cases of acute pancreatitis have been observed during treatment with GLP-1 receptor agonists (see section "Adverse reactions"). Patients should be informed about the characteristic symptoms of acute pancreatitis. If pancreatitis is suspected, treatment with semaglutide should be discontinued; if pancreatitis is confirmed, semaglutide therapy must not be resumed.
Semaglutide should be used with caution when treating patients with a history of pancreatitis.
In the absence of other signs and symptoms of acute pancreatitis, elevated levels of pancreatic enzymes alone are not considered predictive of acute pancreatitis.
Type 2 diabetes mellitus
Semaglutide should not be used as a substitute for insulin in patients with type 2 diabetes mellitus.
Semaglutide should not be used in combination with other GLP-1 receptor agonists. This combination has not been evaluated and is considered likely to increase the risk of adverse reactions related to overdose.
Hypoglycaemia in patients with type 2 diabetes mellitus
It is known that insulin and sulfonylureas can cause hypoglycaemia. The risk of hypoglycaemia may be increased in patients treated with semaglutide in combination with a sulfonylurea or insulin. The risk of hypoglycaemia may be reduced by reducing the dose of sulfonylurea or insulin at the initiation of GLP-1 receptor agonist therapy. The addition of the medicinal product Wegovy FlexTouch to treatment regimens containing insulin has not been evaluated.
Diabetic retinopathy in patients with type 2 diabetes mellitus
An increased risk of complications of diabetic retinopathy has been observed in patients with diabetic retinopathy who received semaglutide (see section "Adverse reactions"). Rapid improvement in glucose control has been associated with transient worsening of diabetic retinopathy; however, the possibility of other mechanisms cannot be excluded. Patients with diabetic retinopathy should be closely monitored and managed according to clinical guidelines. There is no experience with the use of the medicinal product Wegovy FlexTouch in patients with type 2 diabetes mellitus who have uncontrolled or potentially unstable diabetic retinopathy. Use of Wegovy FlexTouch in such patients is not recommended.
Underserved populations
The safety and efficacy of the medicinal product Wegovy FlexTouch have not been studied in the following patient populations:
- patients using other medicinal products for weight control,
- patients with type 1 diabetes mellitus,
- patients with severe renal impairment (see section "Dosage and administration"),
- patients with severe hepatic impairment (see section "Dosage and administration"),
- patients with New York Heart Association (NYHA) Class IV heart failure.
Use in these patients is not recommended.
There is limited experience with the use of the medicinal product Wegovy FlexTouch in the following patient populations:
- patients aged 75 years and older (see section "Dosage and administration"),
- patients with mild or moderate hepatic impairment (see section "Dosage and administration"),
- patients with inflammatory bowel disease,
- patients with diabetic gastroparesis.
Wegovy FlexTouch should be used with caution in these patients.
Sodium content
This medicinal product contains less than 1 mmol sodium (23 mg) per dose and is therefore considered essentially "sodium-free".
Use during pregnancy or breastfeeding.
Women of reproductive potential
Women of reproductive potential are advised to use contraception during treatment with semaglutide (see section "Interaction with other medicinal products and other forms of interaction").
Pregnancy
Animal studies have shown reproductive toxicity (see section "Preclinical safety data"). Data on the use of semaglutide in pregnant women are limited. Therefore, semaglutide should not be used during pregnancy. If a patient is planning pregnancy or becomes pregnant, semaglutide should be discontinued. Due to the long elimination half-life of semaglutide, treatment should be discontinued at least 2 months prior to planned conception (see section "Pharmacokinetics").
Breastfeeding
Semaglutide was excreted in milk in rat lactation studies. A risk to the breastfed infant cannot be excluded. Semaglutide should not be used during breastfeeding.
Fertility
The effect of semaglutide on fertility in humans is unknown. Semaglutide did not affect fertility in male rats. In female rats, prolonged estrous cycle and reduced number of ovulations were observed at doses associated with body weight loss.
Ability to affect the speed of reactions when driving or operating machinery.
Semaglutide has no or negligible influence on the ability to drive or operate machinery. However, dizziness may occur, predominantly during the dose-escalation period. If dizziness occurs, patients should exercise caution when driving or operating machinery.
Patients with type 2 diabetes mellitus
When semaglutide is used in combination with a sulfonylurea or insulin, patients should be advised to take precautions to avoid hypoglycaemia while driving or operating machinery (see section "Special precautions for use").
Method of administration and dosing.
Dosing
Adults
The maintenance dose of semaglutide 2.4 mg once weekly is achieved by starting at a dose of 0.25 mg. To reduce the likelihood of gastrointestinal adverse reactions, the dose should be escalated over a 16-week period to the maintenance dose of 2.4 mg once weekly (see Table 8). If significant gastrointestinal reactions occur, consideration should be given to delaying dose escalation or reducing to the previous dose until symptoms improve. Doses higher than 2.4 mg per week are not recommended.
Table 8. Dose escalation schedule
| Dose escalation |
Weekly dose |
| Week 1–4 |
0.25 mg |
| Week 5–8 |
0.5 mg |
| Week 9–12 |
1 mg |
| Week 13–16 |
1.7 mg |
| Maintenance dose |
2.4 mg |
Children
For children aged 12 years and older, the same dose-escalation schedule as for adults should be used (see Table 8). The dose should be increased up to 2.4 mg (maintenance dose) or until the maximum tolerated dose is reached. Weekly doses above 2.4 mg are not recommended.
Patients with type 2 diabetes
At the initiation of semaglutide in patients with type 2 diabetes, consider reducing the dose of concomitantly administered insulin or insulin secretagogues (such as sulfonylureas) to reduce the risk of hypoglycemia; see section "Special precautions".
Missed dose
If a dose has been missed, it should be administered as soon as possible within 5 days of the missed dose. If more than 5 days have passed, the missed dose should be skipped and the next dose administered on the regularly scheduled day. In this way, patients can resume their once-weekly dosing schedule. If more than one dose has been missed, consider reducing the starting dose when restarting treatment.
Special patient groups
Elderly patients (over 65 years of age)
Dose adjustment based on age is not required. Experience in patients aged ≥ 75 years is limited, and increased sensitivity in some elderly individuals cannot be excluded.
Renal impairment
Dose adjustment is not required in patients with mild or moderate renal impairment. Experience with semaglutide in patients with severe renal impairment is limited. Semaglutide is not recommended in patients with severe renal impairment (eGFR < 30 mL/min/1.73 m²), including patients with end-stage renal disease (see sections "Special precautions", "Adverse reactions", and "Pharmacokinetics").
Hepatic impairment
Dose adjustment is not required in patients with mild or moderate hepatic impairment. Experience with semaglutide in patients with severe hepatic impairment is limited. Semaglutide is not recommended in patients with severe hepatic impairment. In patients with mild or moderate hepatic impairment, semaglutide should be used with caution (see sections "Special precautions" and "Pharmacokinetics").
Administration method
Subcutaneous administration
Wegovy FlexTouch is administered once weekly at any time of day, independent of meals.
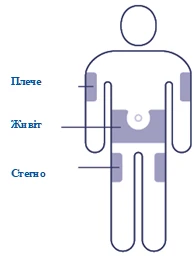
The medication is administered subcutaneously in the abdominal area, thigh, or upper arm. Injection sites may be rotated. The product must not be administered intravenously or intramuscularly.
If necessary, the day of weekly administration may be changed provided that the interval between two doses is at least 3 days (> 72 hours). After selecting a new administration day, continue treatment according to the once-weekly schedule.
When administering a single dose from the pre-filled Wegovy FlexTouch pen, the pen should be firmly pressed against the skin until the yellow line stops moving. The injection takes approximately 5–10 seconds.
Prior to use, patients should be advised to carefully read the Wegovy FlexTouch pen user manual included in the package leaflet.
For additional information before use, see section "Instructions for use of the injection pen".
| Instructions for Using the Wegovy FlexTouch Pen |
|||
| Before starting to use the Wegovy FlexTouch pen once weekly, always read these instructions carefully and speak with your doctor, nurse, or pharmacist about how to properly administer Wegovy FlexTouch. Wegovy FlexTouch is a pre-filled multi-dose pen containing four prescribed doses, corresponding to four weekly uses. Please use the tracking table located inside the lid of the cardboard box to monitor how many doses you have used and how many remain in your pen. Wegovy FlexTouch is available in five different pens, each containing one of the following prescribed doses of semaglutide:
Always begin by checking the label on your pen to ensure it contains the Wegovy FlexTouch dose prescribed for you Your pen is designed for use with single-use 30G, 31G, and 32G needles up to 8 mm in length. The package contains:
|
|||
|
|
|||
|
|||
| Check the name and dose of your pen to confirm it contains the Wegovy FlexTouch dose prescribed for you. Remove the pen cap. (See Figure A). |
|
||
| Ensure the solution in the pen is clear and colourless. Look through the pen window. If the solution is cloudy or discoloured, do not use the pen. (See Figure B). |
|
||
| Always use a new needle for each injection. Take a needle when you are ready to administer the injection. Check that the paper seal and outer needle cap are undamaged, as damage may compromise sterility. If there is any damage, use a new needle. Remove the paper seal. (See Figure C). |
|
||
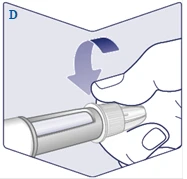
| Screw the needle onto the pen. Turn it to ensure the needle is securely attached. (See Figure D). |
|
||
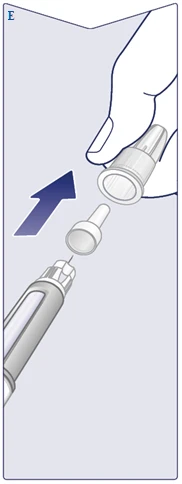
| The needle has two caps. You must remove both caps. If you forget to remove both caps, you will not be able to inject Wegovy FlexTouch. Remove the outer needle cap and keep it. It will be needed after the injection to safely remove the needle from the pen. Remove the inner needle cap and discard it. A drop of solution may appear at the tip of the needle. However, flow must be checked when using a new pen for the first time. See section "Checking the function of each new pen" below. Never use a bent or damaged needle. For more information on handling needles, see section "About your needles" below. (See Figure E). |
|
||
| Checking the function of each new pen |
|||
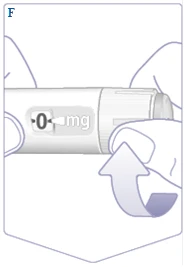
| If you have previously used a Wegovy FlexTouch pen, proceed to section 2. "Setting the dose". Only before the first use of each new Wegovy FlexTouch pen, check the flow. Turn the dose selector until the dose counter displays the flow check symbol (). (See Figure F). |
|
||
| Ensure the flow check symbol aligns with the dose pointer. (See Figure G). |
|
||
| Flow check |
|||
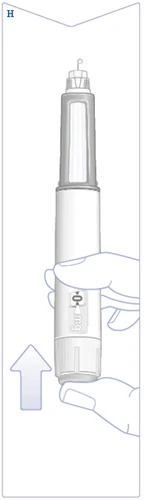
| Hold the pen with the needle pointing upward. Press and hold the injection button until the dose counter returns to the symbol . The symbol must align with the dose pointer. A drop of solution should appear at the needle tip. This drop indicates your pen is ready for use. If no drop appears, repeat the flow check. This should be done only twice. If there is still no drop, replace the needle and repeat the flow check. Do not use the pen if a drop of solution still does not appear. (See Figure H). |
|
||
|
|||
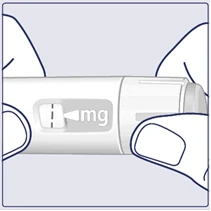
| Turn the dose selector until the dose counter stops and displays your prescribed dose. (See Figure I). |
|
||
| The dotted line () on the dose counter helps identify the dose. The dose selector clicks differently when turned forward, backward, or after a dose. You will hear a click each time you turn the dose selector. Do not set the dose by counting the number of clicks you hear. (See Figure J). |
|
||
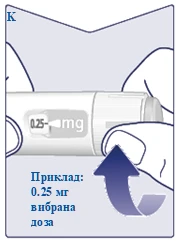
| When your prescribed dose aligns with the dose pointer, your dose is set. In this illustration, dose is shown as an example. If the dose counter stops before you reach your prescribed dose, refer to section "Do you have enough Wegovy FlexTouch?" below. (See Figure K). |
|
||
| Choose the injection site Select a site on the upper arm, thigh, or abdomen (stay at least 5 cm from the navel). You may inject in the same body area each week, but ensure the injection is not in the exact same spot as the previous time. |
|
||
|
|||
| Insert the needle under the skin. Ensure you can see the dose counter. Do not cover the dose counter with your fingers, as this may interrupt the injection. (See Figure L). |
|
||
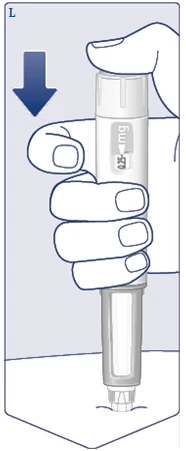
| Press and hold the injection button until the dose counter shows . (See Figure M). Keep the needle under the skin after the dose counter returns to 0, and count slowly to 6. must align with the dose pointer. You may then hear or feel a click as the dose counter returns to . (See Figure N). |
|
||
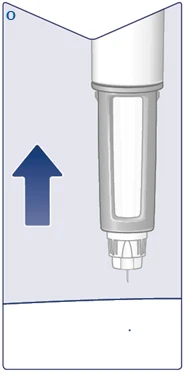
| Remove the needle from the skin. If removed too early, you may see solution leaking from the needle tip, and the full dose will not be delivered. If a drop of blood appears at the injection site, gently press the area to stop bleeding. You may see a drop of solution at the needle tip after injection. This is normal and does not affect the dose delivered. (See Figure O). |
|
||
|
|||
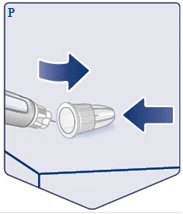
| Place the outer needle cap on a flat surface and replace the outer needle cap over the needle, without touching the needle or the outer needle cap. Once the needle is covered, press firmly and fully onto the outer needle cap. (See Figure P). |
|
||
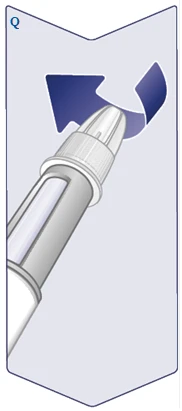
| Unscrew the needle and dispose of it carefully according to instructions from your doctor, nurse, pharmacist, or local authorities. Never attempt to re-sheath the inner needle cap. You may suffer a needlestick injury. Always remove the needle from the pen immediately after each injection to prevent needle blockage, contamination, infection, and inaccurate dosing. Never store the pen with a needle attached. (See Figure Q). |
|
||
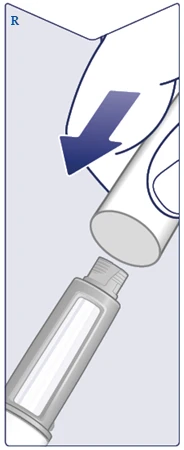
| Replace the pen cap after each use to protect the solution from light. (See Figure R). |
|
||
| When the pen is empty, dispose of the pen without the needle according to instructions from your doctor, nurse, pharmacist, or local authorities. The pen cap and empty box may be discarded with household waste. |
|||
| About your needles |
|||
| How to identify a blocked or damaged needle
What to do with a blocked needle
|
|||
| Caring for your pen |
|||
| Handle your pen carefully. Rough handling or improper use may result in incorrect dosing. If this occurs, you may not achieve the desired effect from this medication.
|
|||
| Do you have enough Wegovy FlexTouch? |
|||
| If the dose counter stops before reaching your prescribed dose, there is insufficient solution to deliver a full dose. Dispose of this pen and use a new Wegovy FlexTouch pen. |
|
||
| Important information |
|||
|
|||








Children
Dose adjustment is not required for children 12 years of age and older. The safety and efficacy of semaglutide in children under 12 years of age have not been established.
Overdose
Overdose with semaglutide may be associated with gastrointestinal disturbances, which could lead to dehydration. In case of overdose, the patient should be monitored for clinical signs, and appropriate supportive treatment should be initiated.
Adverse reactions.
Summary of safety profile
In four phase 3 clinical studies, 2650 patients received the medicinal product Wegovy FlexTouch. The duration of the studies was 68 weeks. The most commonly reported adverse reactions were gastrointestinal disorders, including nausea, diarrhea, constipation, and vomiting.
Adverse Reactions
The adverse reactions listed below were identified in phase 3 clinical trials. The frequency of adverse reactions is based on data from phase 3 clinical trials.
Adverse reactions are classified by system organ class and frequency of occurrence. Frequency was assessed according to the following scale: very common (≥ 1/10), common (≥ 1/100 to < 1/10), uncommon (≥ 1/1,000 to < 1/100), rare (≥ 1/10,000 to < 1/1,000), very rare (< 1/10,000).
Immune system disorders: rare – anaphylactic reaction.
Metabolism and nutrition disorders: common – hypoglycaemia in patients with type 2 diabetes^a.
Nervous system disorders: very common – headache^b; common – dizziness^b.
Eye disorders: common – diabetic retinopathy in patients with type 2 diabetes^a.
Cardiac disorders: uncommon – hypotension, orthostatic hypotension, increased heart rate^a,^c.
Gastrointestinal disorders: very common – vomiting^a,^b, diarrhoea^a,^b, constipation^a,^b, nausea^a,^b, abdominal pain^b,^c; common – gastritis^b,^c, gastroesophageal reflux disease^b, dyspepsia^b, eructation^b, flatulence^b, bloating^b; uncommon – acute pancreatitis^a, delayed gastric emptying.
Hepatobiliary disorders: common – cholelithiasis^a.
Skin and subcutaneous tissue disorders: common – hair loss^a; rare – angioedema.
General disorders and administration site conditions: very common – fatigue^b,^c; common – injection site reactions^c.
Investigations: uncommon – increased amylase levels, increased lipase levels^c.
^a Descriptions of individual adverse reactions are provided below.
^b Mostly observed during dose escalation.
^c Grouped preferred terms.
Description of Individual Adverse Reactions
Gastrointestinal Adverse Reactions
In 68-week studies, nausea occurred in 43.9% of patients treated with semaglutide (16.1% in the placebo group), diarrhoea in 29.7% (15.9% in the placebo group), and vomiting in 24.5% (6.3% in the placebo group). Most cases were mild to moderate in severity and transient. Constipation occurred in 24.2% of patients receiving semaglutide (11.1% in the placebo group) and was mild or moderate in severity but more prolonged. In patients receiving semaglutide, the median duration of nausea was 8 days, vomiting 2 days, diarrhoea 3 days, and constipation 47 days.
Patients with moderate renal impairment (eGFR ≥ 30 mL/min/1.73 m²) may experience more pronounced gastrointestinal effects during treatment with semaglutide.
Gastrointestinal reactions led to permanent discontinuation of treatment in 4.3% of patients.
Acute Pancreatitis
The incidence of expert-confirmed acute pancreatitis during phase 3 clinical trials was 0.2% with semaglutide and < 0.1% with placebo.
Acute Gallstone Disease/Cholelithiasis
Cholelithiasis was reported in 1.6% of patients and led to cholecystitis in 0.6% of patients receiving semaglutide. Cholelithiasis and cholecystitis were observed in 1.1% and 0.3% of patients receiving placebo, respectively.
Hair Loss
Hair loss was reported in 2.5% of patients receiving semaglutide and 1.0% of patients receiving placebo. The reaction was predominantly mild in severity, and most patients recovered while continuing treatment. Hair loss was more frequently observed in patients with greater weight loss (≥ 20%).
Increased Heart Rate
In phase 3 clinical trials, patients receiving semaglutide experienced a mean increase in heart rate of 3 beats per minute (bpm) from a baseline mean of 72 bpm. The proportion of patients with an increase in heart rate of ≥ 10 bpm from baseline at any time during the treatment period was 67.0% in the semaglutide group compared to 50.1% in the placebo group.
Immunogenicity
Due to the potential immunogenic properties of protein- or peptide-containing medicinal products, patients treated with semaglutide may develop antibodies. The percentage of patients with a positive test for anti-semaglutide antibodies at any time after treatment initiation was low (2.9%), and no patient had neutralising antibodies to semaglutide or antibodies with neutralising GLP-1 effect by the end of clinical trials. High concentrations of semaglutide during treatment may have reduced assay sensitivity, so the risk of false-negative results cannot be excluded. However, in patients with positive antibody tests during and after treatment, the presence of antibodies was transient and had no apparent impact on efficacy or safety.
Hypoglycaemia in Patients with Type 2 Diabetes
In the STEP 2 trial, clinically significant hypoglycaemia occurred in 6.2% (0.1 events per patient-year) of patients receiving semaglutide compared to 2.5% (0.03 events per patient-year) of patients receiving placebo. Hypoglycaemia with semaglutide occurred both with and without concomitant use of sulphonylureas. One episode (0.2% of patients, 0.002 events per patient-year) was classified as severe in a patient not receiving concomitant sulphonylurea. The risk of hypoglycaemia increased when semaglutide was used with sulphonylureas.
Diabetic Retinopathy Complications
In a 2-year clinical trial investigating semaglutide 0.5 mg and 1 mg versus placebo, 3,297 patients with type 2 diabetes, high cardiovascular risk, long-standing diabetes, and inadequate glycaemic control were enrolled. In this trial, events of diabetic retinopathy complications occurred in more patients receiving semaglutide (3.0%) than in those receiving placebo (1.8%). This was observed in patients with confirmed diabetic retinopathy who were receiving insulin. The difference between treatment groups emerged early and persisted throughout the trial. In the STEP 2 trial, eye disorders were reported in 6.9% of patients receiving Wegovy FlexTouch, 6.2% of patients receiving semaglutide 1 mg, and 4.2% of patients receiving placebo. Most cases were reported as diabetic retinopathy (4.0%, 2.7%, and 2.7%, respectively) and non-proliferative retinopathy (0.7%, 0%, and 0%, respectively).
Paediatric Population
In a clinical trial involving children aged 12 to 18 years with obesity or overweight and at least one weight-related comorbidity, 133 patients received Wegovy FlexTouch. The trial duration was 68 weeks.
Overall, the frequency, type, and severity of adverse reactions in children were similar to those in adults. Cholelithiasis was reported in 3.8% of patients receiving Wegovy FlexTouch and 0% of patients receiving placebo.
After 68 weeks of treatment, no impact on growth or pubertal development was observed.
Reporting of Adverse Reactions
Reporting of adverse reactions after marketing authorization is important. It allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals, pharmacists, patients, or their legal representatives should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at: https://aisf.dec.gov.ua.
Shelf Life. 3 years.
After first use: 6 weeks. Store below 30 °C or in the refrigerator (at 2 °C – 8 °C).
Storage Conditions.
Store in the refrigerator (2 °C – 8 °C). Keep away from freezing elements. Do not freeze.
Store the pre-filled pen with the cap attached to protect from light.
Keep out of the reach of children.
Incompatibilities.
This medicinal product must not be mixed with other medicinal products, as compatibility studies have not been conducted.
Packaging.
0.25, 0.5 mg
A 1.5 mL glass cartridge (Type I glass), closed at one end with a rubber (chlorobutyl) stopper and at the other end with an aluminium cap with a laminated rubber liner (bromobutyl/polyisoprene). The cartridge is contained in a single-use pre-filled pen made of polypropylene, polyoxymethylene, polycarbonate, and acrylonitrile butadiene styrene.
1 pre-filled pen and 4 single-use NovoFine® Plus needles in a cardboard box.
1 mg, 1.7 mg
A 3 mL glass cartridge (Type I glass), closed at one end with a rubber (chlorobutyl) stopper and at the other end with an aluminium cap with a laminated rubber liner (bromobutyl/polyisoprene). The cartridge is contained in a single-use pre-filled pen made of polypropylene, polyoxymethylene, polycarbonate, and acrylonitrile butadiene styrene.
1 pre-filled pen and 4 single-use NovoFine® Plus needles in a cardboard box.
2.4 mg
A 3 mL glass cartridge (Type I glass), closed at one end with a rubber (chlorobutyl) stopper and at the other end with an aluminium cap with a laminated rubber liner (bromobutyl/polyisoprene). The cartridge is contained in a single-use pre-filled pen made of polypropylene, polyoxymethylene, polycarbonate, and acrylonitrile butadiene styrene.
1 or 3 pre-filled pens and 4 or 12 single-use NovoFine® Plus needles in a cardboard box.
Prescription Category. Prescription only.
Marketing Authorisation Holder/Manufacturer.
A/T Novo Nordisk.
Address of the Marketing Authorisation Holder/Manufacturer and Location of its Operations.
Novo Allé
2880 Bagsværd
Denmark.