Rixubis
UkraineTable of Contents
INSTRUCTIONS FOR MEDICAL USE OF THE MEDICINAL PRODUCT RIXUBIS
Composition:
Active substance: nonacog gamma*;
1 vial contains:
250 IU** of nonacog gamma, recombinant human coagulation factor IX (rDNA), corresponding to a concentration of 50 IU/mL after reconstitution of the product with 5 mL of solvent;
500 IU** of nonacog gamma, recombinant human coagulation factor IX (rDNA), corresponding to a concentration of 100 IU/mL after reconstitution of the product with 5 mL of solvent;
1000 IU** of nonacog gamma, recombinant human coagulation factor IX (rDNA), corresponding to a concentration of 200 IU/mL after reconstitution of the product with 5 mL of solvent;
2000 IU** of nonacog gamma, recombinant human coagulation factor IX (rDNA), corresponding to a concentration of 400 IU/mL after reconstitution of the product with 5 mL of solvent;
3000 IU** of nonacog gamma, recombinant human coagulation factor IX (rDNA), corresponding to a concentration of 600 IU/mL after reconstitution of the product with 5 mL of solvent.
Excipients: L-histidine, sodium chloride, calcium chloride, mannitol, sucrose, polysorbate 80.
1 vial of solvent contains: water for injections – 5 mL.
_________________________________________________________________________
* Nonacog gamma (recombinant blood coagulation factor IX (rDNA)) is a purified single-chain glycoprotein containing 415 amino acids. It is produced using recombinant DNA technology in a Chinese hamster ovary cell line.
** Activity (IU) is determined by a one-stage clotting factor assay according to the European Pharmacopoeia. The specific activity of RIXUBIS is approximately 200–390 IU/mg protein.
Pharmaceutical form. Powder and solvent for solution for injection.
Main physicochemical properties: white or almost white powder; solvent – clear, colorless solution.
Pharmacotherapeutic group. Antihemorrhagics. Coagulation factor IX.
ATC code B02BD04.
Pharmacological properties.
Pharmacodynamics.
Mechanism of action
RIXUBIS contains recombinant coagulation factor IX (nonacog gamma). Factor IX is a single-chain glycoprotein with a molecular weight of approximately 68,000 daltons. It is a vitamin K-dependent clotting factor produced in the liver. Factor IX is activated by factor XIa in the intrinsic coagulation pathway and by the factor VII/tissue factor complex in the extrinsic coagulation pathway. Activated factor IX, together with activated factor VIII, activates factor X. Activated factor X converts prothrombin into thrombin. Thrombin then converts fibrinogen into fibrin, resulting in clot formation.
Pharmacodynamics
Hemophilia B is a sex-linked inherited disorder of the blood coagulation system caused by reduced levels of factor IX, leading to severe bleeding into joints, muscles, or internal organs, either spontaneously or following trauma or surgery. Replacement therapy allows for an increase in factor IX plasma levels and thus provides temporary correction of the deficiency, reducing the tendency to bleed.
Clinical efficacy and safety
Bleeding prevention and control in previously treated patients aged 12 years and older
The efficacy of RIXUBIS was evaluated in the uncontrolled part of a combined open-label phase 1/3 study, in which 73 previously treated patients aged 12–59 years received RIXUBIS for prophylaxis and/or on-demand treatment of bleeding episodes. All patients had severe (factor IX level < 1%) or moderate (factor IX level < 2%) hemophilia B. Of these, 59 patients received RIXUBIS for prophylaxis. Data from 56 patients who received RIXUBIS for at least 3 months were included in the efficacy analysis population. An additional 14 patients received RIXUBIS only for on-demand treatment of bleeding episodes. Patients in the on-demand treatment cohort were required to have had at least 12 documented bleeding episodes requiring treatment during the 12 months prior to enrollment. The mean duration of treatment in the on-demand cohort was 3.5 ± 1.00 months (median 3.4; range 1.2 to 5.1 months). The mean annualized bleeding rate (ABR) was 33.9 ± 17.37, with a median of 27.0 and a range of 12.9 to 73.1.
The median ABR during prophylactic treatment with RIXUBIS was 2.0 for all bleeds, 0.0 for spontaneous bleeds, and 0.0 for joint bleeds. In 24 patients (42.9%), no bleeding episodes occurred.
A total of 249 bleeding episodes were treated with RIXUBIS, including 197 joint bleeds and 52 non-joint bleeds (in soft tissues, muscles, body cavities, intracranial space, and others). Of the 249 bleeding episodes, 163 were moderate in severity, 71 were mild, and 15 were severe. Treatment was individualized based on the severity, cause, and location of each bleed. The majority (211 episodes; 84.7%) of the 249 bleeding episodes were resolved with 1–2 infusions. Hemostatic efficacy in stopping bleeding was rated as excellent or good in 96% of all treated bleeding episodes.
Bleeding prevention and control in previously treated patients under 12 years of age
The efficacy of RIXUBIS was evaluated in a combined phase 2/3 study involving 23 male patients aged 1.8–11.8 years (median age: 7.10 years), of whom 11 were under 6 years of age. All patients had previously received treatment and were administered RIXUBIS for prophylaxis and control of bleeding episodes. All patients had severe (factor IX level < 1%) or moderate (factor IX level < 2%) hemophilia B. All 23 patients received prophylactic treatment with RIXUBIS for at least 3 months and were included in the efficacy analysis.
The median ABR was 2.0, with a median of 0.0 for spontaneous bleeds and 0.0 for joint bleeds.
Nine patients (39.1%) experienced no bleeding episodes.
A total of 26 bleeding episodes were treated with RIXUBIS, of which 23 were trauma-related, 2 were spontaneous, and 1 was of unknown origin. Nineteen bleeds occurred outside joints (in soft tissues, muscles, body cavities, intracranial space, and others), and 7 were joint bleeds, one of which was a target joint bleed. Of the 26 bleeding episodes, 15 were mild, 9 were moderate, and 2 were severe. Treatment was individualized based on severity, cause, and location of the bleed. The majority (23 episodes; 88.5%) were treated with 1–2 infusions. Hemostatic efficacy in stopping bleeding was rated as excellent or good in 96.2% of all treated bleeding episodes.
Perioperative management
The safety and efficacy of RIXUBIS in the perioperative setting were evaluated in a multicenter, prospective, open-label, uncontrolled phase 3 study involving previously treated males with severe and moderate hemophilia B. The per-protocol efficacy analysis included 37 surgical procedures—comprising major and minor surgeries, dental procedures, or other surgical invasive interventions—performed in 27 patients aged 17 to 57 years. Twenty procedures were classified as major, including 13 orthopedic and 3 dental surgeries. Seventeen procedures, including 10 tooth extractions, were considered minor. Patients undergoing major surgery were required to undergo pharmacokinetic (PK) assessment. All patients received dosing based on individual stepwise recovery data for factor IX activity. The recommended initial dose of RIXUBIS was intended to maintain factor IX activity levels at 80–100% during major surgery and at 30–60% during minor procedures. RIXUBIS was administered as a bolus intravenous infusion.
Hemostasis was maintained throughout the entire study period.
Pharmacokinetics
Previously treated patients aged 12 years and older
A randomized, double-blind, controlled crossover pharmacokinetic study of RIXUBIS and a comparator product was conducted in males without active bleeding (aged ≥15 years) as part of a central combined phase 1/3 study. Patients received either product as a single intravenous infusion. The mean (± standard deviation) and median dose of RIXUBIS in the per-protocol analysis population (n = 25) was 74.69 ± 2.37 and 74.25 IU/kg, respectively, with a range of 71.27 to 79.38 IU/kg. Pharmacokinetic parameters were calculated based on measurements of factor IX activity in blood samples collected over a 72-hour period following each infusion.
Pharmacokinetic assessment of RIXUBIS was repeated in an open-label, uncontrolled study involving males who had participated in the initial crossover PK study and who had received prophylactic treatment with RIXUBIS for 26 ± 1 weeks (mean ± standard deviation), with a cumulative exposure to RIXUBIS of at least 30 days. The dose range of RIXUBIS used in the repeat PK study was 64.48 to 79.18 IU/kg (n = 23).
Table 1 presents the pharmacokinetic parameters in all evaluable patients (per-protocol analysis).
Table 1
| Parameter |
RIXUBIS Initial crossover study (N = 25) |
RIXUBIS Repeat assessment (N = 23) |
| AUC0-72 h (MU • h/dL)a Mean ± standard deviation (SD) Median (range) |
1067.81 ± 238.42 1108.35 (696.07–1571.16) |
1156.15 ± 259.44 1170.26 (753.85–1626.81) |
| In vivo recovery at Cmax (MU/dL : MU/kg)b Mean ± SD Median (range) |
0.87 ± 0.22 0.88 (0.53–1.35) |
0.95 ± 0.25 0.93 (0.52–1.38) |
| Half-life (h) Mean ± SD Median (range) |
26.70 ± 9.55 24.58 (15.83–52.34) |
25.36 ± 6.86 24.59 (16.24–42.20) |
| Cmax (MU/dL) Mean ± SD Median (range) |
66.22 ± 15.80 68.10 (41.70–100.30) |
72.75 ± 19.73 72.40 (38.50–106.30) |
| Mean residence time (h) Mean ± SD Median (range) |
30.82 ± 7.26 28.93 (22.25–47.78) |
29.88 ± 4.16 29.04 (21.32–37.52) |
| Vssc (dL/kg) Mean ± SD Median (range) |
2.02 ± 0.77 1.72 (1.10–3.94) |
1.79 ± 0.45 1.74 (1.12–2.72) |
| Clearance (dL/kg•h) Mean ± SD Median (range) |
0.0644 ± 0.0133 0.0622 (0.0426–0.0912) |
0.0602 ± 0.0146 0.0576 (0.0413–0.0945) |
a Area under the plasma concentration–time curve from 0 to 72 hours after infusion.
b Calculated as the ratio of Cmax of factor IX activity prior to treatment initiation divided by the dose in IU/kg, where Cmax is the maximum measured factor IX activity level after infusion.
c Volume of distribution at steady state.
Incremental recovery of factor IX activity level, measured 30 minutes after infusion, was determined in all patients enrolled in the combined phase 1/3 study on day 1 of treatment, at physician visits on weeks 5, 13, and 26, and at the time of study completion or discontinuation, if it did not coincide with the week 26 visit. The obtained data indicate that the incremental recovery remained stable throughout the entire observation period (see Table 2).
Table 2
| Parameter |
Day 1 of treatment (N = 73) |
Week 5 (N = 71) |
Week 13 (N = 68) |
Week 26 (N = 55) |
End of study/ study discontinuation day b (N = 23) |
| Increase in recovery activity 30 min after infusion (U/dL : U/kg) a Mean ± SD Median (range) |
0.79 ± 0.20 0.78 (0.26–1.35) |
0.83 ± 0.21 0.79 (0.46–1.48) |
0.85 ± 0.25 0.83 (0.14–1.47) |
0.89 ± 0.12 0.88 (0.52–1.29) |
0.87 ± 0.20 0.89 (0.52–1.32) |
a Calculated as the (factor IX level at 30 minutes post-infusion) divided by the dose in IU/kg, where C30 min is the measurement of factor IX activity 30 minutes after infusion.
b If not aligned with the visit time at Week 26.
Pediatric population (previously treated patients, under 12 years of age)
In a combined Phase 2/3 study, initial pharmacokinetic assessment of RIXUBIS was performed in 23 pediatric male patients who were bleeding-free. To minimize discomfort from frequent blood sampling for each individual patient, patients were randomized into two groups with different blood sampling schedules. The mean dose (± standard deviation) and median dose of RIXUBIS in the full analysis set (n = 23) were 75.50 ± 3.016 and 75.25 IU/kg, respectively, with a range of 70.0 to 83.6 IU/kg. Pharmacokinetic parameters were estimated based on measurements of factor IX activity in blood samples collected over a period of up to 72 hours after infusion.
Table 3 presents the pharmacokinetic parameters for all patients (full analysis set).
Table 3
| Parameter |
Up to 6 years (N = 11) |
From 6 to 12 years (N = 12) |
All (N = 23) |
| AUCinf (MO • h/dL)а Mean ± SD Median (range) |
723.7 ± 119.00 717.2 (488–947) |
886.0 ± 133.66 863.7 (730–1138) |
808.4 ± 149.14 802.9 (488–1138) |
| Half-life (h) Mean ± SD Median (range) |
27.67 ± 2.66 27.28 (24.0–32.2) |
23.15 ± 1.58 22.65 (21.8–27.4) |
25.31 ± 3.13 24.48 (21.8–32.2) |
| Mean residence time (h) Mean ± SD Median (range) |
30.62 ± 3.27 30.08 (26.2–36.2) |
25.31 ± 1.83 24.74 (23.7–30.3) |
27.85 ± 3.73 26.77 (23.7–36.2) |
| Vssb (dL/kg) Mean ± SD Median (range) |
3.22 ± 0.52 3.16 (2.65–4.42) |
2.21 ± 0.32 2.185 (1.70–2.70) |
2.7 ± 0.67 2.69 (1.70–4.42) |
| Clearance (dL/kg • h) Mean ± SD Median (range) |
0.1058 ± 0.01650 0.1050 (0.081–0.144) |
0.0874 ± 0.01213 0.0863 (0.069–0.108) |
0.0962 ± 0.01689 0.0935 (0.069–0.144) |
a Area under the plasma concentration-time curve from 0 hours to infinity.
b Steady-state volume of distribution.
The incremental recovery of Factor IX activity level at 0.5 hours after infusion was determined in all patients enrolled in the combined Phase 2/3 study, at the time of initial pharmacokinetic assessment (Day 1 of treatment), at physician visits on Weeks 5, 13, and 26, and at the time of study completion or discontinuation, if it did not coincide with the Week 26 visit. The obtained data indicate that the incremental recovery remained stable over time across all pediatric age groups (see Tables 4, 5, and 6 below).
Table 4
Incremental recovery of Factor IX activity 0.5 hours after RIXUBIS infusion in pediatric patients across both age groups
| Recovery of activity 30 minutes after infusion |
PK (Day 1 of treatment) All (N = 22) |
Week 5 All (N = 23) |
Week 13 All (N = 21) |
Week 26 All (N = 21) |
| (U/dL: U/kg)a Mean ± SD Median (range) |
0.67 ± 0.16 0.69 (0.31–1.00) |
0.68 ± 0.12 0.66 (0.48–0.92) |
0.71 ± 0.13 0.66 (0.51–1.00) |
0.72 ± 0.15 0.734 (0.51–1.01) |
a Calculated as the (30-minute Factor IX level prior to treatment initiation) divided by the dose in IU/kg, where C30 min is the measurement of Factor IX activity 30 minutes after infusion.
Table 5
Incremental recovery of activity following administration of RIXUBIS 30 minutes after infusion in pediatric patients under 6 years of age
| Recovery of activity 30 minutes after infusion |
PK (Day 1 of treatment) All (N = 10) |
Week 5, All (N = 11) |
Week 13, All (N = 10) |
Week 26, All (N = 10) |
| (U/dL: U/kg)a Mean ± SD Median (range) |
0.59 ± 0.13 0.59 (0.31–0.75) |
0.63 ± 0.10 0.6 (0.49–0.80) |
0.68 ± 0.12 0.66 (0.51–0.84) |
0.65 ± 0.13 0.61 (0.51–0.84) |
a Calculated as the ratio of the factor IX activity level at 30 minutes post-infusion (C30 min) to the dose administered in IU/kg, where C30 min is the measured factor IX activity level at 30 minutes after infusion.
Table 6
Incremental recovery of factor IX activity 30 minutes after infusion of RIXUBIS in pediatric patients aged 6 to 12 years
| Activity recovery increment 30 minutes after infusion |
PK (Day 1 of treatment) All (N = 12) |
Week 5, All (N = 12) |
Week 13, All (N = 11) |
Week 26, All (N = 11) |
| (U/dL: U/kg)a Mean ± SD Median (range) |
0.73 ± 0.16 0.71 (0.51–1.00) |
0.73 ± 0.13 0.70 (0.48–0.92) |
0.73 ± 0.14 0.70 (0.54–1.00) |
0.8 ± 0.14 0.78 (0.56–1.01) |
a Calculated as the (30-minute factor IX level prior to treatment) divided by the dose in IU/kg, where C30 min is the measurement of factor IX activity 30 minutes after infusion.
Clinical characteristics.
Indications.
For the treatment and prevention of bleeding in patients with hemophilia B (congenital factor IX deficiency).
RIXUBIS is indicated for patients of all age groups.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients listed in the section "Composition".
Known allergic reaction to hamster proteins.
Interaction with other medicinal products and other forms of interaction.
No interactions have been reported between medicinal products based on recombinant human blood coagulation factor IX (rDNA) and other medicinal products.
Special precautions for use
Traceability
To improve traceability of biological medicinal products, the name and batch number of the administered product should be clearly documented, for example in the patient's diary.
Hypersensitivity
Allergic-type hypersensitivity reactions have been reported with the use of RIXUBIS. The medicinal product contains residual amounts of hamster proteins. If symptoms of hypersensitivity occur, patients or caregivers should be advised to discontinue the medicinal product immediately and contact their physician. Patients should be informed about early signs of hypersensitivity reactions, including urticaria, generalized urticaria, chest tightness, wheezing, hypotension, and anaphylaxis.
The risk is highest during initial treatment with factor IX concentrates in previously untreated patients, particularly in patients with a high risk of gene mutations. Publications have reported an association between the development of factor IX inhibitors and allergic reactions, especially in patients with a high risk of gene mutations. Therefore, patients experiencing allergic reactions should be evaluated for the presence of such inhibitors.
In the event of shock, standard anti-shock treatment should be administered.
Inhibitors
After repeated treatment with medicinal products based on recombinant human coagulation factor IX (rDNA), patients should be monitored for the development of neutralizing antibodies (inhibitors). The quantity of inhibitors should be determined in Bethesda units (BU) using an appropriate biological assay method.
Publications have reported an association between the development of factor IX inhibitors and allergic reactions. Therefore, patients experiencing allergic reactions should be evaluated for the presence of such inhibitors. Patients who have developed factor IX inhibitors are at increased risk of anaphylaxis upon subsequent treatment with factor IX.
Due to the risk of allergic reactions with factor IX concentrates, initial administration of factor IX should, at the physician’s discretion, be performed under medical supervision to ensure appropriate medical care can be provided in case of allergic reactions.
Nephrotic syndrome
Cases of nephrotic syndrome have been reported following immune tolerance induction in patients with hemophilia B who had previously developed factor IX inhibitors.
Thromboembolism
Due to the potential risk of thrombotic complications, clinical monitoring for early signs of thrombosis and consumptive coagulopathy, including appropriate biological assays, should be implemented when administering this medicinal product to patients with liver disease, postoperative patients, neonates, or patients at risk of thrombotic events or disseminated intravascular coagulation (DIC). In each of these cases, the benefits of treatment with RIXUBIS should be carefully weighed against the risk of such complications.
Cardiovascular diseases
In patients with pre-existing cardiovascular diseases, replacement therapy with factor IX may increase the risk of cardiovascular pathology.
Complications related to catheter use
When using a central venous access device (CVAD), potential complications related to CVAD use should be considered, including local infections, bacteremia, and thrombosis at the catheter site.
Considerations regarding excipients
After reconstitution, this medicinal product contains less than 1 mmol (23 mg) of sodium per vial, i.e., it is practically sodium-free. Depending on the patient’s body weight and prescribed dosage, patients may receive more than one vial of RIXUBIS, which should be taken into account when following a sodium-controlled diet.
Elderly patients
Patients aged 65 years and older were not included in clinical studies with RIXUBIS. It is unknown whether their response to treatment differs from that of younger patients. As with all other patients, dose selection for elderly patients should be individualized.
Children
The above warnings and precautions apply to the treatment of both adults and children.
Use during pregnancy or breastfeeding
Pregnancy
Data on the use of factor IX in pregnant women are lacking or limited. Reproductive toxicity studies with factor IX have not been conducted in animals.
Factor IX should be used during pregnancy and breastfeeding only if clearly indicated.
Breastfeeding
There are no data on the excretion of factor IX or its metabolites in human breast milk.
Fertility
There is no information on the effect of factor IX on fertility.
Ability to drive and use machines
RIXUBIS does not affect the ability to drive or operate machinery.
Method of Administration and Dosage
Treatment should be carried out under the supervision of a physician experienced in the management of hemophilia.
Monitoring During Treatment
During treatment, appropriate monitoring of factor IX levels is recommended to guide dosing and the frequency of repeat infusions. Individual patient response to factor IX may vary, with differences in half-life and recovery levels. Patients with low or excessive body weight may require dose adjustments based on body weight. During major surgical procedures, precise monitoring during replacement therapy through coagulation factor assays (factor IX activity in blood plasma) is essential.
To ensure achievement of desired factor IX plasma activity levels, careful monitoring using an appropriate factor IX activity assay is recommended, with corresponding adjustments in dose and frequency of repeat infusions as necessary. When using a one-stage in vitro coagulation factor assay based on activated partial thromboplastin time (aPTT) to determine factor IX activity in patient plasma samples, the results may significantly depend on both the type of aPTT reagent and the reference standard used. This is particularly important when changing laboratories and/or reagents used for testing.
Dosage
The required dose and duration of replacement therapy depend on the severity of factor IX deficiency, the location and severity of bleeding, as well as the patient's clinical condition, age, and pharmacokinetic parameters of factor IX, such as the increment in factor IX activity level and half-life.
The administered amount of factor IX is expressed in International Units (IU) according to the current WHO standard for factor IX-containing medicinal products. Factor IX activity in blood plasma is expressed either as a percentage (relative to the normal human plasma value) or in International Units (relative to the International Standard for factor IX in plasma).
One International Unit (IU) of factor IX activity corresponds to the activity level of factor IX in 1 ml of normal human plasma.
Adults
On-demand Treatment
The required dose of factor IX for patients aged 12 years and older is calculated based on empirical data indicating that administration of 1 International Unit (IU) of factor IX per 1 kg of body weight increases factor IX activity in plasma by 0.9 IU/dL (range: 0.5 to 1.4 IU/dL) or by 0.9% of normal activity.
The required dose is determined using the formula below.
| Required number of units |
= |
body weight (kg) |
× |
desired increase in factor IX level (% or BU/dL) |
× |
reciprocal of the observed factor IX recovery value (dL/kg) |
For a stepwise increase in the restoration of activity level by 0.9 IU/dl per 1 IU/kg, the dose is calculated using the following formula:
| Required number of units |
= |
body weight (kg) |
× |
desired increase in factor IX level (% or IU/dL) |
× |
1.1 dL/kg |
The dose amount and frequency of scheduled administration should always be directed toward ensuring clinical efficacy in each individual patient.
In the case of the hemorrhagic events listed below, Factor IX activity levels should not be lower than the specified plasma activity level (% of normal or IU/dL) for the appropriate period of time. Table 7 can be used to determine dosing for bleeding episodes and surgical interventions.
Table 7
| Severity of bleeding/ type of surgical intervention |
Required factor IX level, % or IU/dl |
Dosing frequency (hours)/ duration of treatment (days) |
| Bleeding Early hemarthrosis, muscle bleeds, or oral cavity hemorrhage |
20–40 |
Repeat every 24 hours. For at least 1 day, until bleeding stops, as indicated by pain resolution, or until wound healing. |
| More pronounced hemarthrosis, muscle bleeds, or hematoma |
30–60 |
Repeat infusion every 24 hours for 3–4 days or longer, until pain subsides and significant functional impairment resolves. |
| Life-threatening bleeding |
60–100 |
Repeat infusion every 8–24 hours until the threat is resolved. |
| Surgical procedures Minor surgical procedures, including tooth extraction |
30–60 |
Every 24 hours, for at least 1 day, until wound healing. |
| Major surgical procedures |
80–100 (before and after surgery) |
Repeat infusion every 8–24 hours until adequate wound healing; thereafter continue treatment for at least 7 additional days to maintain factor IX activity levels between 30% and 60% (IU/dl). |
Careful monitoring of replacement therapy is particularly important in the case of major surgical intervention or life-threatening bleeding.
Prophylaxis
For long-term prophylaxis of bleeding in patients aged 12 years and older with severe haemophilia B, doses of 40–60 IU of factor IX per kg body weight administered every 3–4 days are generally used. In some cases, depending on pharmacokinetics, age, bleeding phenotype, and level of physical activity, it may be necessary to shorten the intervals between administrations or to increase the dose.
Continuous infusion
Do not administer RIXUBIS by continuous infusion.
Method of administration
Intravenous administration.
If the patient or a caregiver is to administer the product, they must be properly trained in performing this procedure.
RIXUBIS should be administered at a rate comfortable for the patient, up to a maximum of 10 ml/min.
After reconstitution, the solution is clear, colourless, free from foreign particulate matter, with a pH of 6.8–7.2. The osmolarity is greater than 240 mOsmol/kg.
Only plastic syringes with a Luer-lock tip should be used for administration of this product.
RIXUBIS is administered intravenously after reconstitution of the powder with the solvent provided.
- Use only the solvent and reconstitution device (BAXJECT II) supplied in the package for reconstituting the product.
- A syringe with a Luer-lock tip must be used for administration.
- Do not use the BAXJECT II device if it is damaged, if the sterile protection system or packaging is damaged, or if there are any signs of deterioration.
Reconstitution
Use aseptic techniques.
- If the medicinal product has been stored in the refrigerator, remove the RIXUBIS powder vial and the solvent vial from the refrigerator and allow them to warm to room temperature (15 °C to 30 °C).
- Wash hands thoroughly with warm water and soap.
- Remove the caps from the powder and solvent vials.
- Wipe the stoppers with alcohol swabs. Place the vials on a clean, flat surface.
- Open the BAXJECT II device package by peeling off the paper lid, taking care not to touch the inner surface of the device (Figure a). Do not remove the device from the package.
- Turn the package upside down and attach the transparent plastic spike to the solvent vial stopper. Holding the package by its edges, remove the package from the BAXJECT II device (Figure b). Do not remove the blue cap from the BAXJECT II device.
- With the BAXJECT II device now attached to the solvent vial, invert the system so that the solvent vial is above the device. Attach the white plastic spike to the stopper of the RIXUBIS vial. Under vacuum, the solvent will transfer into the vial containing RIXUBIS (Figure c).
- Gently mix until the powder is completely dissolved. The product dissolves rapidly (within 2 minutes). Ensure that RIXUBIS is completely dissolved, otherwise not all of the reconstituted product will pass through the filter in the device. Reconstituted medicinal products should be inspected visually for particulate matter and discoloration prior to administration. The solution should be clear or slightly opalescent. Do not use solutions that are cloudy or contain precipitate.
| Figure a |
Figure b |
Figure c |
|
|
||
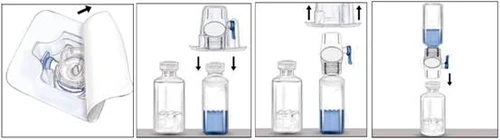
Do not refrigerate the medicinal product after reconstitution. Use it immediately after preparation.
Administration
Use aseptic techniques.
- Remove the blue cap from the BAXJECT II device. Take care not to draw air into the syringe. Attach the syringe to the BAXJECT II device (Figure d).
- Invert the system (the vial with the reconstituted solution should be on top). Fill the syringe with the reconstituted solution by slowly pulling back the plunger (Figure e).
- Detach the syringe.
- Attach a butterfly needle to the syringe. Administer the solution intravenously. The solution should be administered slowly at a rate comfortable for the patient, no faster than 10 ml per minute.
| Figure d |
Figure e |
|
|
|
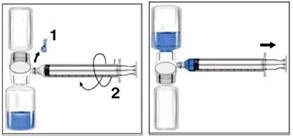
Always record the name and batch number of RIXUBIS when using the product (e.g., in your personal diary) to keep a record of the medicinal products you have used.
Any unused medicinal product or waste material must be disposed of in accordance with local requirements.
The chemical and physical in-use stability of the diluted, ready-to-use medicinal product is 3 hours at a temperature not exceeding 30 °C. From a microbiological standpoint, despite the dilution method preventing microbial contamination, the medicinal product should be used immediately. If not used immediately, the duration and conditions of storage are the responsibility of the user. Do not refrigerate.
Children
Patients aged 12 to 17 years
The dosing is the same for adults and children aged 12 to 17 years.
Patients under 12 years of age
On-demand treatment
The required dose of factor IX for patients under 12 years of age is calculated based on empirical findings indicating that administration of one international unit (IU) of factor IX per 1 kg of body weight increases factor IX activity in blood plasma by 0.7 IU/dL (range from 0.31 to 1.0 IU/dL) or by 0.7% of normal activity.
The required dose is determined using the formula below.
| Required number of units |
= |
body weight (kg) |
× |
desired increase in factor IX level (% or IU/dL) |
× |
reciprocal of the observed recovery value (dL/kg) |
For a stepwise increase in the recovery of activity level by 0.7 IU/dl per 1 IU/kg, the dose is calculated using the following formula:
| Required number of units |
= |
body weight (kg) |
× |
desired increase in factor IX level (% or IU/dL) |
× |
1.4 dL/kg |
For determining the dose during bleeding episodes and surgical interventions, the same table as for adults can be used (see Table 7 above).
Prophylaxis
The recommended dose range for treating pediatric patients under 12 years of age is 40 to 80 IU/kg every 3–4 days. In some cases, depending on pharmacokinetics, age, bleeding phenotype, and level of physical activity, it may be necessary to shorten the dosing intervals or increase the dose.
Overdose.
Effects of doses higher than recommended for RIXUBIS have not been described.
Adverse reactions
Rare cases of hypersensitivity or allergic reactions (which may include angioneurotic edema, burning and itching sensations at the infusion site, chills, facial flushing, generalized urticaria, headache, rash, hypotension, lethargy, nausea, restlessness, tachycardia, chest tightness, tinnitus, vomiting, wheezing) have been observed. These reactions may occasionally progress to severe anaphylaxis (including shock), occurring in close temporal association with the administration of factor IX inhibitors.
Cases of nephrotic syndrome have been reported in patients with hemophilia B and a history of allergic reactions who underwent immune tolerance induction therapy in the presence of factor IX inhibitors.
Very rare cases of antibody formation against hamster proteins, associated with the occurrence of hypersensitivity reactions, have been observed.
In patients with hemophilia B, neutralizing antibodies (inhibitors) to factor IX may develop, which manifest as suboptimal treatment response. In such cases, consultation with a specialized hemophilia treatment center is recommended.
The use of factor IX-containing medicinal products carries a potential risk of thromboembolic events, with a higher risk associated with the use of low-purity purified products. Administration of low-purity factor IX products has been associated with cases of myocardial infarction, disseminated intravascular coagulation, venous thrombosis, and pulmonary embolism. Such adverse reactions are rarely observed with highly purified factor IX products.
List of adverse reactions in tabular form
A total of 99 individuals were enrolled in clinical studies involving the use of RIXUBIS, with at least one dose administered, resulting in a total of 5 reported adverse reactions. The table below presents information on adverse reactions classified by system organ classes (SOC) according to the MedDRA classification (using preferred terms).
The frequency of adverse reactions is categorized as follows: very common (> 1/10), common (from > 1/100 to < 1/10), uncommon (from > 1/1000 to < 1/100), rare (from > 1/10,000 to < 1/1000), very rare (< 1/10,000), frequency not known (based on available data, frequency cannot be estimated).
Within each frequency category, adverse reactions are listed in descending order of severity.
Table 8
| Adverse reactions reported in clinical trials and spontaneous reports |
||
| System organ class by MedDRA classification |
Adverse reactions |
Frequency per patient |
| Immune system disorders |
Hypersensitivity a) |
Unknown |
| Nervous system disorders |
Disturbance of taste |
Common |
| Musculoskeletal and connective tissue disorders |
Limb pain |
Common |
a) ADR (adverse reaction) is explained below in the section.
Description of individual adverse reactions
Hypersensitivity
Allergic-type reactions manifested as dyspnea, pruritus, generalized urticaria, and rash.
Children
The frequency, type, and severity of adverse reactions in children are expected to be the same as in adults. However, data regarding untreated patients are lacking, since only previously treated patients were included in clinical trials. Therefore, immunogenicity studies regarding inhibitor development in patients in this risk group were not conducted.
Reporting of suspected adverse reactions
Reporting of suspected adverse reactions after drug registration is of great importance. It allows monitoring of the benefit-risk balance of this medicinal product. Healthcare professionals and patients or their legal representatives should report all cases of suspected adverse reactions and lack of efficacy through the Automated Information System for Pharmacovigilance at the following link: https://aisf.dec.gov.ua.
Shelf life.
3 years.
Storage conditions.
Store at a temperature not exceeding 30 °C. Do not freeze.
Keep out of reach of children.
Incompatibility.
Due to the lack of compatibility studies, this medicinal product must not be mixed with other medicinal products.
Only plastic syringes with a Luer tip may be used when administering this medicinal product. Due to the adsorption of human coagulation factor IX onto the internal surfaces of certain infusion equipment, the administered dose of the drug may be inaccurate.
Packaging.
1 vial with powder (250 IU, 500 IU, 1000 IU, 1500 IU, 2000 IU, or 3000 IU) and 1 vial with solvent (5 ml of water for injections) and 1 BAXJECT II reconstitution device per carton.
Prescription category.
Prescription only.
Manufacturer.
Baxalta Belgium Manufacturing SA / Baxalta Belgium Manufacturing SA.
Manufacturers' addresses and locations of their business activities.
Boulevard Rene Branquart 80, Lessines, 7860, Belgium / Boulevard Rene Branquart 80, Lessines, 7860, Belgium.