Refacto af
UkraineTable of Contents
INSTRUCTIONS for medical use of the medicinal product ReFacto AF (ReFacto® AF)
Composition:
Active substance: moroctocog alfa (recombinant blood coagulation factor VIII);
1 vial contains 250 IU or 500 IU, or 1000 IU, or 2000 IU of moroctocog alfa;
1 pre-filled syringe contains 3000 IU of moroctocog alfa;
Excipients: sucrose, calcium chloride dihydrate, L-histidine, polysorbate 80, sodium chloride.
Solvent: sodium chloride, water for injections.
Pharmaceutical form.
Lyophilisate for solution for injection.
Main physicochemical properties:
presentation form – vial:
white lyophilisate, practically free from clearly visible foreign particles, moisture, and defects in vial stoppering;
solvent: colorless clear solution, practically free from visible inclusions;
presentation form – pre-filled syringe:
in the upper chamber – white lyophilisate, practically free from clearly visible foreign particles, moisture, and defects in container stoppering;
in the lower chamber – solvent: colorless clear solution, practically free from visible inclusions.
Pharmacotherapeutic group.
Antihemorrhagics. Blood coagulation factor VIII. ATC code B02B D02.
Pharmacological properties.
Pharmacodynamics.
ReFacto AF contains recombinant blood coagulation factor VIII with the B-domain deleted (moroctocog alfa). The potency of the medicinal product is expressed in International Units (IU) determined by a chromogenic assay according to the European Pharmacopoeia. The specific activity of ReFacto AF is 7,600–13,800 IU/mg protein.
Moroctocog alfa is a glycoprotein with an approximate molecular weight of 170,000 Da, composed of 1438 amino acids. Its amino acid sequence is similar to the 90 + 80 kDa form of factor VIII (i.e., with the B-domain deleted), and its post-translational modifications are comparable to those found in plasma-derived factor VIII molecules.
Moroctocog alfa is produced using recombinant DNA technology in Chinese hamster ovary cells. The manufacturing process of ReFacto AF has been modified to eliminate the possibility of contamination with exogenous human or animal proteins during cell culture, purification, or final product manufacturing. Concurrently, the product name was changed from ReFacto to ReFacto AF.
The functional characteristics of ReFacto AF are similar to those of endogenous factor VIII. Factor VIII activity is significantly reduced in patients with hemophilia A, necessitating replacement therapy.
Upon intravenous administration to patients with hemophilia, the infused factor VIII binds to circulating von Willebrand factor.
Activated factor VIII acts as a cofactor for activated factor IX, accelerating the conversion of factor X to activated factor X. Activated factor X then converts prothrombin into thrombin. Thrombin, in turn, converts fibrinogen into fibrin, leading to clot formation. Hemophilia A is an X-linked inherited bleeding disorder caused by reduced levels of factor VIII:C, resulting in severe hemorrhages into joints, muscles, or internal organs. Bleeding episodes may occur spontaneously or following trauma or surgery. Replacement therapy increases plasma factor VIII levels, thereby temporarily correcting the deficiency and reducing the tendency to bleed.
Clinical efficacy
The data presented in Table 1 were obtained from clinical studies of ReFacto AF administered to previously untreated patients (PUPs) and previously treated patients (PTPs) under 12 years of age.
Table 1
Dosing and efficacy in pediatric patients
| Parameter |
PLP |
PLP |
PNP |
| Dosing by body weight (IU/kg) for prophylactic infusiona |
N = 14 (28, 51) |
N = 13 (21, 49) |
N = 22 (17, 161) |
| Total ABR among all subjects, |
-- |
-- |
N = 23 (0.0; 39.5) |
| Total ABR for subjects who were on-demand at baselinec, |
N = 5 (1.6; 50.6) |
N = 9 (0.0; 46.6) |
-- |
| Total ABR for subjects who were on prophylaxis at baselinec, |
N = 13 (0.0; 11.2) |
N = 9 (0.0; 13.0) |
-- |
| Dosing by body weight (IU/kg) per bleeding episode for treatment of bleeding, median (min., max.) |
N = 13 (28; 86) |
N = 14 (17; 229) |
N = 21 (11; 221) |
| % of bleeds successfully treated with ≤ 2 infusions |
98.7% |
98.8% |
96.7% |
a The dosage and frequency of Refacto AF administration in the study were determined by the physician based on local treatment standards.
b Subjects in the PUP study were not required to adhere to regular continuous prophylactic treatment; however, with the exception of one subject (who received only on-demand treatment), most subjects received regular prophylactic infusions. Several subjects entered the study receiving on-demand treatment and subsequently switched to prophylactic treatment, while some subjects received only sporadic prophylactic infusions.
c At the beginning of the study, subjects in the PTP study reported their treatment regimen as factor VIII (prophylaxis or on-demand) and were not required to maintain this regimen as a condition of study participation. The dosage and frequency of Refacto AF administration in the study were determined by the physician based on local treatment standards.
Abbreviation: ABR – annual bleeding rate.
It should be noted that ABR values are not comparable across different factor concentrates or across different clinical studies.
Immune tolerance induction.
Data on immune tolerance induction were collected in patients with hemophilia A who developed factor VIII inhibitors. Within the pivotal clinical study of Refacto involving 25 previously untreated patients, data on immune tolerance induction were collected (15 with high titer, 10 with low titer). Of these 25 patients, inhibitor titers decreased to < 0.6 BU (Bethesda units) in 20 patients, including 11 of the initial 15 who had high titers (≥ 5 BU/mL) and 9 of the 10 who had low titers. A similar decrease in inhibitor titers was observed in 5 of 6 patients who had low inhibitor titers but did not receive immune tolerance induction treatment. Long-term follow-up was not performed.
Pharmacokinetics.
Pharmacokinetic properties of Refacto, determined in a crossover study of Refacto and plasma-derived factor VIII concentrate involving 18 previously treated patients (PTPs), are shown in Table 2. Quantitative assays were performed using a chromogenic substrate method (see section "Posology and method of administration").
Table 2
| Pharmacokinetic parameter assessment of ReFacto in patients with hemophilia A |
|||
| Parameter |
Mean |
SD |
Median |
| AUCt (IU·h/mL) |
19.9 |
4.9 |
19.9 |
| t½ (h) |
14.8 |
5.6 |
12.7 |
| CL (mL/h·kg) |
2.4 |
0.75 |
2.3 |
| mean residence time (h) |
20.2 |
7.4 |
18.0 |
| recovery (IU/dL increase in factor VIII:C per IU/kg administered factor VIII) |
2.4 |
0.38 |
2.5 |
Abbreviations: AUCt – area under the plasma concentration-time curve from zero to the last measured concentration; t½ – elimination half-life; CL – clearance; SD – standard deviation.
In a study comparing the activity of ReFacto AF, ReFacto, and factor VIII in plasma of patients using a chromogenic assay, bioequivalence of ReFacto AF and ReFacto was established. The ratios of least-squares mean pharmacokinetic parameters of ReFacto AF to ReFacto were 100.6%, 99.5%, and 98.1% for recovery, AUCt, and AUC∞ (area under the plasma concentration-time curve from zero to infinity), respectively. The corresponding 90% confidence intervals for the ratios of least-squares mean pharmacokinetic parameters of ReFacto AF to ReFacto were within the bioequivalence window (80–125%), indicating bioequivalence between ReFacto AF and ReFacto.
In a crossover pharmacokinetic study, pharmacokinetic parameters of ReFacto AF were determined in 25 previously treated patients (≥12 years of age) at baseline and after 6 months of repeated administration of ReFacto AF. The ratios of least-squares mean pharmacokinetic parameters after 6 months of treatment compared to baseline values were 107%, 100%, and 104% for recovery, AUCt, and AUC∞, respectively. The corresponding 90% confidence intervals for the ratios of pharmacokinetic parameters after 6 months compared to baseline were within the equivalence range (80–125%), indicating absence of time-dependent changes in the pharmacokinetic properties of ReFacto AF.
In the same study, where the activity of ReFacto AF, recombinant full-length factor VIII (comparator product), and factor VIII activity were measured in plasma samples from patients (30 previously treated patients, ≥12 years of age) using a one-stage clotting assay, ReFacto AF demonstrated pharmacokinetic bioequivalence to the comparator product when applying the standard approach for establishing bioequivalence.
In previously untreated patients, pharmacokinetic parameters of ReFacto were assessed using a chromogenic assay. In these patients (n = 59; median age 10 ± 8.3 months), mean recovery at week "0" was 1.5 ± 0.6 IU/dL per IU/kg (range: 0.2–2.8 IU/dL per IU/kg), which is lower than values obtained in previously treated patients at week "0" with a mean recovery of 2.4 ± 0.4 IU/dL per IU/kg (range: 1.1–3.8 IU/dL per IU/kg). In previously untreated patients, mean recovery remained stable over a 2-year period (5 visits during this time) and ranged between 1.5–1.8 IU/dL per IU/kg. A population pharmacokinetic modeling study, including data from 44 previously untreated patients, showed that the predicted mean elimination half-life was 8.0 ± 2.2 hours.
In a study evaluating ReFacto AF in 19 PUPs, recovery levels in 17 children aged from 28 days to 2 years were 1.32 ± 0.65 IU/dL per IU/kg, and in 2 children aged 2 to <6 years were 1.7 and 1.8 IU/dL per IU/kg. Except for cases where inhibitors were detected, mean recovery levels remained stable over time (6 visits over a 2-year period), with individual values ranging from 0 (in presence of inhibitor) to 2.7 IU/dL per IU/kg. Table 3 presents the observed pharmacokinetic parameters of ReFacto AF after administration of a 50 IU/kg dose in a study involving 37 previously treated children.
Table 3
| Mean values ± SD of pharmacokinetic parameters of factor VIII after administration of a single 50 IU/kg dose in children (PUP) |
||
| Parameter |
Number of subjects |
Mean valuea ± SD |
| Incremental recovery, IU/dL per IU/kg Under 6 years of age 6 to 12 years of age |
17 19 |
1.7 ± 0.4 2.1 ± 0.8 |
| Cmax, IU/mLb |
19 |
0.9 (45) |
| AUCinf, IU∙h/mLb |
14 |
9.9 (41) |
| t½, hb |
14 |
9.1 ± 1.9 |
| CL, mL/h/kgb |
14 |
4.4 (30) |
| Vss, mL/kgb |
14 |
56.4 (15) |
| a Geometric mean (geometric CV%) for all parameters except arithmetic mean ± SD for incremental recovery and t½. b Patients aged 6 to 12 years only. Abbreviations: Cmax – maximum observed plasma concentration; CV – coefficient of variation; AUCinf – area under the plasma concentration-time curve from 0 to infinity; t½ – elimination half-life; CL – clearance; SD – standard deviation; Vss – volume of distribution at steady state. |
||
Clinical characteristics.
Indications.
Treatment and prevention of bleeding episodes in patients with hemophilia A (congenital deficiency of blood coagulation factor VIII).
ReFacto AF is indicated for use in adults and children of all ages, including infants.
ReFacto AF does not contain von Willebrand factor and therefore is not indicated for the treatment of von Willebrand disease.
Contraindications.
Hypersensitivity to the active substance or to any of the excipients.
Known allergic reaction to hamster proteins.
Interaction with other medicinal products and other forms of interaction.
No interactions between recombinant human blood coagulation factor VIII and other medicinal products have been reported.
Special precautions for use.
Tracking
To improve tracking of biological medicinal products, the name and batch number of the administered product should be clearly documented. For this purpose, patients may attach one of the detachable adhesive labels from the vial or prefilled syringe to their personal diary to record the batch number, or use it for reporting any adverse effects.
Hypersensitivity
ReFacto AF may cause hypersensitivity reactions of an allergic type. The product contains trace amounts of hamster proteins. Patients should be advised to discontinue use of the product immediately and seek medical advice if symptoms of hypersensitivity occur. Patients should be informed about early signs of hypersensitivity reactions, including urticaria, generalized urticaria, chest tightness, wheezing, hypotension, and anaphylaxis.
In the event of shock, standard medical treatment for shock should be initiated.
Inhibitors
Development of neutralizing antibodies (inhibitors) to factor VIII is a known complication in the treatment of patients with hemophilia A. These are usually IgG immunoglobulins directed against the procoagulant activity of factor VIII. Their presence in plasma is determined by a modified quantitative method and expressed in Bethesda units (BU) per 1 mL. The risk of inhibitor formation correlates with the severity of the disease and exposure to factor VIII, being highest during the first 50 exposure days to the product, but persists throughout life, although it is infrequent.
The clinical significance of inhibitor formation depends on their titer, with low-titer inhibitors posing a lower risk of inadequate clinical response compared to high-titer inhibitors.
Patients receiving treatment with recombinant factor VIII coagulation factors should be closely monitored for inhibitor development through appropriate clinical observation and laboratory testing. If expected plasma factor VIII activity levels are not achieved or if bleeding is not controlled with an appropriate dose of the product, assays should be performed to determine the presence of factor VIII inhibitors. In patients with high inhibitor levels, factor VIII therapy may be ineffective, and alternative treatment options should be considered. Management of such patients should be performed by physicians experienced in the treatment of hemophilia and factor VIII inhibitor management.
Reporting of inadequate efficacy
In clinical studies and during post-marketing use, cases of inadequate efficacy of ReFacto have been reported, primarily in patients using the product for prophylaxis. Lack of efficacy has been described as bleeding into target joints, bleeding into new joints, or a subjective sensation by the patient of the onset of a new bleed. When prescribing ReFacto AF, it is important to individualize dosing and monitor factor levels in each patient to ensure an adequate therapeutic response (see section "Adverse Reactions").
Cardiovascular complications
In patients with existing risk factors for cardiovascular complications, replacement therapy with factor VIII may increase cardiovascular risks.
Catheter-related complications
If a central venous access device is required, the risks associated with its use, including local infections, bacteremia, and catheter thrombosis, should be considered (see section "Adverse Reactions").
Sodium content
After reconstitution, one vial or prefilled syringe contains 1.27 mmol (or 29 mg) of sodium, equivalent to 1.5% of the WHO recommended maximum daily intake of sodium for adults (2 g). Depending on the patient's body weight and dosage of ReFacto AF, patients may use several vials or prefilled syringes. This information should be taken into account for patients on a sodium-restricted diet.
Use during pregnancy or breastfeeding.
Studies on the effect of factor VIII on reproductive function in animals have not been conducted; therefore, data on its effect on fertility are lacking. Since hemophilia A is rare in women, experience with the use of factor VIII during pregnancy and breastfeeding is limited. Therefore, factor VIII should be used during pregnancy and breastfeeding only if clearly needed.
Ability to affect reaction speed when driving or operating machinery.
ReFacto AF does not affect the ability to drive or operate machinery.
Administration and Dosage.
Treatment should be initiated under the supervision of a physician experienced in the management of hemophilia A.
Monitoring of Treatment.
To determine the dose required during the course of treatment and the frequency of repeat infusions, monitoring of factor VIII levels is recommended. The response to factor VIII administration may vary among individual patients, resulting in different levels of recovery and different half-lives. The dose calculated based on body weight may need adjustment in patients with excessive or low body weight. In particular, careful monitoring of replacement therapy using coagulation assays (factor VIII activity in plasma) is mandatory during major surgical procedures.
For monitoring factor VIII activity levels in patients during treatment with Refacto AF, the chromogenic assay method is recommended. If in vitro activated partial thromboplastin time (aPTT) is used to determine factor VIII activity in patient blood samples via a one-stage coagulation activity assay, the results of factor VIII activity may be influenced by both the types of reagents and the reference standards used in quantification. There may also be significant differences between quantitative results obtained using the chromogenic assay method and those obtained using the aPTT one-stage coagulation activity assay. Typically, results obtained by the one-stage coagulation activity assay are 20–50% lower than those obtained using the chromogenic assay method. To correct for this discrepancy, a Refacto AF laboratory standard may be used (see section "Pharmacokinetics"). This is particularly important when different laboratories and/or reagents are employed.
Dosage.
The dosage and duration of replacement therapy depend on the severity of factor VIII deficiency, the location and severity of bleeding, and the patient's clinical condition. Doses used should be adjusted according to the patient's clinical response. Higher doses of the product or specific appropriate treatment may be required in the presence of inhibitors.
The quantity of factor VIII units administered is expressed in International Units (IU), referenced to the WHO standard for factor VIII-containing products. Factor VIII activity in blood plasma is expressed as a percentage (relative to its concentration in normal human plasma) or in International Units (relative to the international standard for factor VIII in plasma). One IU of factor VIII activity is equivalent to the amount of factor VIII present in 1 mL of normal human plasma.
The activity of another moroctocog alfa product (Xyntha), registered outside Ukraine, is determined using a manufacturing standard calibrated against the WHO international standard using a one-stage coagulation activity assay. Due to differences in the methods used to determine the activity of Xyntha and Refacto AF, 1 IU of Xyntha (calibrated using a one-stage assay) is approximately equivalent to 1.38 IU of Refacto AF (calibrated using a chromogenic assay). If a patient normally receiving Xyntha is prescribed Refacto AF, the physician may consider adjusting the dose based on factor VIII recovery levels.
Patients with hemophilia A should be advised to carry an adequate supply of factor VIII product according to their prescribed treatment regimen in case treatment becomes necessary during travel. Patients should be advised to consult their physician prior to travel.
On-demand Treatment.
The calculation of the required factor VIII dose is based on empirical data indicating that 1 IU of factor VIII per 1 kg of body weight increases factor VIII activity in blood plasma by 2 IU/dL. The required dose is calculated using the following formula:
Required number of units (IU) =
body weight (kg) × desired increase in factor VIII level (%, or IU/dL) × 0.5 (IU/kg per IU/dL),
where 0.5 IU/kg per IU/dL represents the reciprocal of the recovery typically observed after factor VIII infusion.
The dose and frequency of administration of the prescribed product should always be individually determined based on the clinical efficacy of the product.
In the event of hemorrhagic episodes, factor VIII activity in blood plasma should not fall below the levels (in % or IU/dL) specified in Table 4. This table may be used as a guideline for factor VIII dosing in bleeding episodes and surgical interventions.
Table 4
| Severity of bleeding/ type of surgical procedure |
Required factor VIII level (% of normal, or IU/dL plasma) |
Dosing frequency (hours)/ duration of therapy (days) |
| Bleeding |
||
| Early signs of hemarthrosis, muscle hematomas, or oral bleeding |
20–40 |
Repeat administration every 12–24 hours for at least 1 day until bleeding stops, as indicated by absence of pain or evidence of healing |
| Moderate hemarthrosis, muscle bleeds, or hematomas |
30–60 |
Repeat administration every 12–24 hours for 3–4 days or longer until resolution of pain and restoration of joint mobility |
| Life-threatening bleeding |
60–100 |
Repeat administration every 8–24 hours until the life-threatening condition is resolved |
| Surgical procedures |
||
| Minor surgical procedures, including tooth extraction |
30–60 |
Repeat administration every 24 hours for at least 1 day until wound healing |
| Major surgical procedures |
80–100 (before and after surgery) |
Repeat administration every 8–24 hours until adequate wound healing; continue therapy for at least 7 days thereafter to maintain factor VIII activity at 30–60 IU/dL |
Prophylaxis
For long-term prophylaxis of bleeding episodes in patients with severe haemophilia A, the usual regimen is 20–40 IU of factor VIII per kg body weight every 2–3 days. In some cases, particularly for younger patients, higher doses or more frequent administration may be required.
Children
When treating younger paediatric patients (up to 6 years of age) with Refacto AF, higher doses may be required compared to adult patients and older children (see section "Pharmacokinetics").
Elderly patients
Patients aged 65 years and older were not included in clinical studies. In general, dosage for elderly patients should be individually adjusted.
Patients with renal or hepatic impairment
Dosage adjustment for patients with renal or hepatic impairment has not been studied during clinical trials.
Method of administration
Intravenous administration.
Refacto AF is administered by intravenous infusion over several minutes after reconstitution with the appropriate diluent. The infusion rate should be based on patient comfort. Individuals who self-administer the product, but are not healthcare professionals, should receive appropriate training.
Form of release – vial
The lyophilised powder is reconstituted with the appropriate diluent (0.9% sodium chloride solution) supplied in a pre-filled syringe, using a sterile adapter. After adding the diluent, the vial should be gently rotated until the powder is completely dissolved.
More detailed information on the preparation and administration of the solution is provided below.
After reconstitution, the solution is drawn back into the syringe. The solution should be clear or slightly opalescent and colourless. The solution must be discarded if particulate matter or discoloration is observed.
Form of release – pre-filled syringe:
The lyophilised powder in the upper chamber of the pre-filled syringe must be reconstituted with the diluent (sodium chloride solution, 9 mg/mL (0.9%)) located in the lower chamber. The pre-filled syringe should be gently rotated until all powder is dissolved. More detailed information on the preparation and administration of the solution is provided below.
After reconstitution, the solution will be clear or slightly opalescent and colourless. The solution must be discarded if visible particulate matter or discoloration is observed.
Refacto AF, after reconstitution, contains polysorbate 80, which may accelerate the extraction of di-(2-ethylhexyl) phthalate from polyvinyl chloride (PVC). This property should be considered during preparation and administration of the product, including the storage time in a PVC container after reconstitution. It is important to strictly follow the recommendations provided in the section "Storage conditions".
Any unused portions of the medicinal product or materials used for reconstitution and administration must be disposed of in accordance with local requirements.
Form of release – vial
Preparation of the solution:
- Allow the temperature of the lyophilisate and the diluent in the pre-filled syringe to reach room temperature.
- Remove the plastic flip-top cap from the Refacto AF vial to expose the central portion of the rubber stopper.
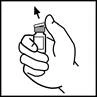
- Wipe the top of the vial with the alcohol swab provided in the package, or use another antiseptic solution, and allow it to dry. After cleaning, do not touch the rubber stopper with hands and avoid contact with any surfaces.
- Peel back the protective covering of the clear plastic packaging of the vial adapter. Do not remove the adapter from the packaging.
- Place the vial on a flat surface. Holding the packaging, place the adapter onto the vial. Press firmly until the adapter clicks into place on the top of the vial and the adapter's spike penetrates the vial's stopper.
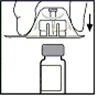
- Remove the packaging from the adapter and dispose of it.
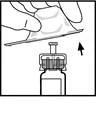
- Attach the plunger rod to the syringe containing the diluent: insert the rod into the stopper of the syringe, press firmly and rotate the rod until it is securely engaged in the stopper.
- Break off the plastic cap with tamper-evident seal from the diluent syringe by breaking the perforation on the cap. This is done by wiggling the cap until the perforation breaks. Do not touch the inner surface of the cap or the tip of the syringe. It may be necessary to replace the cap (if the prepared Refacto AF is not used immediately), so set it aside with the opening facing upwards.
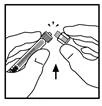
- Place the vial on a flat surface. Attach the syringe with diluent to the vial adapter by inserting the syringe tip into the adapter opening, pressing firmly and rotating the syringe clockwise until a secure connection is achieved.
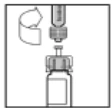
- Slowly press the plunger rod to transfer the entire diluent into the Refacto AF vial.
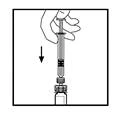
- Leaving the syringe attached to the adapter, gently rotate the vial until the powder is completely dissolved.
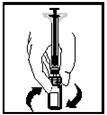
- Before administration, visually inspect the prepared solution for the presence of particulate matter. The resulting solution should be clear or slightly opalescent and colourless.
If more than one Refacto AF vial is used per infusion, each vial should be prepared according to the above instructions. Then, remove the diluent syringe, leaving the vial adapter in place, and a single large syringe with a Luer lock can be used to withdraw the solution from each vial.
- After ensuring the plunger remains fully inserted into the syringe barrel, invert the vial. Slowly withdraw the entire solution through the vial adapter into the syringe.
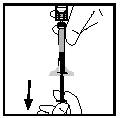
- Disconnect the syringe from the vial adapter by carefully pulling and rotating the syringe counterclockwise. Discard the vial with the attached adapter.
If the solution is not used immediately, the syringe cap should be carefully replaced. Do not touch the syringe tip or the inner surface of the cap.
The prepared solution should be used immediately or within 3 hours after preparation. The prepared solution may be stored at a temperature not exceeding 25 °C until administration.
Administration (intravenous infusion)
Refacto AF should be administered using the infusion set provided and the pre-filled diluent syringe provided, or a single sterile disposable plastic syringe with a Luer lock.
- Attach the syringe to the Luer lock connector of the infusion set.
- Apply a tourniquet and prepare the injection site by thoroughly wiping the skin with the alcohol swab provided in the kit.
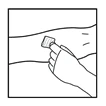
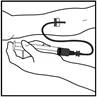
- Insert the infusion set catheter needle into the vein and remove the tourniquet. Remove air from the infusion set catheter by pulling back on the syringe. The prepared product should be administered intravenously over several minutes. The physician may adjust the recommended infusion rate to make the infusion procedure more comfortable for the patient.
Form of release – pre-filled syringe
Preparation of the solution:
- Allow the pre-filled syringe to reach room temperature.
- Retrieve the pre-filled syringe kit for Refacto AF and place it on a clean surface, ensuring all necessary components are present.
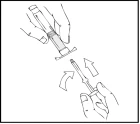
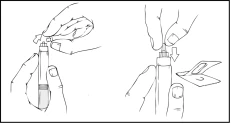
- Hold the plunger rod as shown in the diagram below. Firmly screw the plunger rod into the hole in the finger grip of the Refacto AF pre-filled syringe by pressing and turning clockwise until resistance is felt (approximately 2 turns).
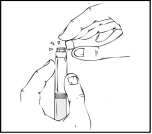
Throughout the solution preparation process, the Refacto AF pre-filled syringe must be held vertically (the white powder should be above the clear diluent) to prevent potential leakage.
- While holding the pre-filled syringe vertically, break off the white protective cap with tamper-evident seal by bending it from right to left (or with gentle rocking motions) to break the cap's perforation and expose the grey rubber cap of the pre-filled syringe tip.
- Remove the blue protective vented sterile cap from its packaging.
While continuously holding the Refacto AF pre-filled syringe vertically, remove the grey rubber cap and replace it with the blue protective vented cap. This protective cap has small openings that allow air to escape, preventing excessive pressure. Do not touch the open end of the syringe or the blue protective cap.
- Carefully and slowly advance the plunger rod, pressing until the two internal plungers within the pre-filled syringe meet and all the diluent has transferred into the upper chamber containing the Refacto AF powder.
Note: To prevent leakage from the syringe tip, do not press the plunger rod with excessive force.
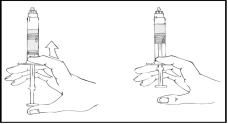
- Holding the Refacto AF syringe vertically, gently rotate it until the powder is dissolved.
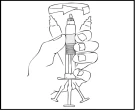
Visually inspect the solution for particulate matter or discoloration. The solution should be clear, slightly opalescent, and colourless. If the solution in the pre-filled syringe contains particulate matter or has changed colour, do not use it.
- While continuing to hold the Refacto AF pre-filled syringe vertically, slowly advance the plunger rod until most, but not all, of the air is expelled from the (upper) chamber.
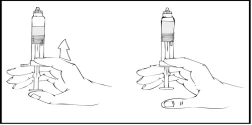
Refacto AF should be administered within 3 hours after reconstitution or after removal of the grey rubber cap from the pre-filled syringe.
If the Refacto AF solution is not used immediately, the syringe should be stored vertically with the blue protective cap on the pre-filled syringe until infusion is ready to be performed. The prepared solution may be stored at room temperature for up to 3 hours. If not used within 3 hours, the solution must be discarded.
Administration (intravenous infusion)
Refacto AF should be administered using the infusion set provided.
- Remove the blue protective cap and securely attach the infusion set to the Refacto AF pre-filled syringe.
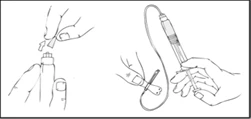
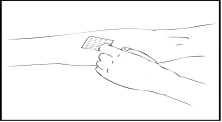
- Apply a tourniquet and prepare the injection site by thoroughly wiping the skin with the alcohol swab provided in the kit.
- Remove the needle protective cap and insert the infusion set catheter needle into the vein. Remove the tourniquet. The prepared Refacto AF solution should be administered intravenously over several minutes. The physician may adjust the recommended infusion rate to make the infusion procedure more comfortable for you. Do not attempt to perform the infusion yourself without proper training.
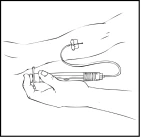
The prepared Refacto AF solution must not be administered through the same infusion set or container as other medicinal products.
- After infusion, remove and discard the infusion set. The residual amount of product remaining in the infusion set does not affect treatment. Additional information on the use of multiple Refacto AF syringes with 10 mL or larger Luer-Lok syringes (10 mL or larger Luer-Lok syringes are not included in the package)
- Prepare the solution in all Refacto AF syringes according to the reconstitution instructions above.
While holding the filled Refacto AF syringe vertically, slowly advance the plunger rod until most, but not all, of the air is expelled from the syringe.
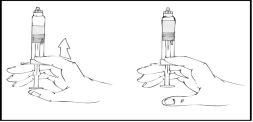
- Unpack the Luer-Lok syringe connector (not included in the package).
- Connect a sterile 10 mL or larger Luer-Lok syringe to one port of the syringe connector, and the Refacto AF syringe to the other open port on the opposite side.
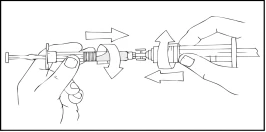
- Holding the Refacto AF syringe on top, slowly press the plunger rod until the contents transfer into the empty 10 mL or larger Luer-Lok syringe.
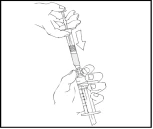
- Remove the empty Refacto AF pre-filled syringe and repeat steps 3 and 4 with additional syringes containing the prepared solution.
- Remove the syringe connector from the 10 mL or larger Luer-Lok syringe and attach the infusion set as described above in the administration instructions.
Unused solution, empty vial(s), and used needles and syringes must be disposed of in an appropriate medical waste container, as these materials may cause harm to others if not properly disposed of.
Children
Refacto AF can be used in children of any age, including infants, as stated in the section "Method of administration and dosage".
Overdose
No symptoms of overdose have been reported with the use of recombinant factor VIII products.
Side effects.
Summary of safety profile
Hypersensitivity and allergic reactions (which may include angioedema, burning and stinging at the infusion site, chills, flushing, generalized urticaria, headache, urticaria, arterial hypotension, lethargy, nausea, restlessness, tachycardia, chest tightness, tinnitus, vomiting, wheezing) occur rarely with the use of Refacto AF and in some cases may progress to severe anaphylaxis, including shock (see section "Special precautions").
Refacto AF may contain trace amounts of hamster protein. Very rarely, antibodies to hamster protein have been observed, but without clinical complications. In a Refacto study, 20 out of 113 (18%) previously treated patients (PTPs) showed increased titers of antibodies to Chinese hamster ovary protein, without apparent clinical effect.
Development of neutralizing antibodies (inhibitors) may occur in patients with hemophilia A who are treated with factor VIII, including Refacto AF. Inhibitor development may manifest as inadequate clinical response. In such cases, consultation with a specialized hemophilia treatment center is recommended.
List of side effects
The information below follows the system organ class classification according to the Medical Dictionary for Regulatory Activities (MedDRA). Adverse reactions are classified by frequency: very common (≥ 1/10); common (≥ 1/100 to < 1/10); and uncommon (≥ 1/1000 to < 1/100). The adverse reactions listed below were observed in clinical studies of Refacto or Refacto AF. Frequencies are based on all reported adverse events occurring during treatment in all clinical trials involving 715 patients.
Within each group, adverse effects are listed in decreasing order of severity.
Blood and lymphatic system disorders:
very common: factor VIII inhibition (in previously untreated patients)*;
uncommon: factor VIII inhibition (in previously treated patients)*.
Immune system disorders: uncommon: anaphylactic reactions.
Metabolism and nutrition disorders: common: decreased appetite.
Nervous system disorders: very common: headache;
common: dizziness;
uncommon: peripheral neuropathy, somnolence, dysgeusia.
Cardiac disorders: uncommon: angina pectoris, tachycardia, palpitations.
Vascular disorders: common: bleeding/hematoma;
uncommon: hypotension, thrombophlebitis, facial hyperemia.
Respiratory, thoracic and mediastinal disorders: very common: cough;
uncommon: dyspnea.
Gastrointestinal disorders: common: diarrhea, nausea, abdominal pain, vomiting.
Skin and subcutaneous tissue disorders: common: urticaria, pruritus, rash;
uncommon: hyperhidrosis.
Musculoskeletal and connective tissue disorders: very common: arthralgia;
common: myalgia.
General disorders and administration site conditions: very common: pyrexia;
common: chills, complications related to permanent venous catheter;
uncommon: asthenia, pain, inflammation, and other reactions at the injection site.
Investigations:
common: positive antibody test; positive test for antibodies to factor VIII;
uncommon: increased blood levels of aspartate aminotransferase, alanine aminotransferase, bilirubin, creatine phosphokinase.
* Frequency is based on studies of all factor VIII products involving patients with severe hemophilia A. PTPs – previously treated patients; PUPs – previously untreated patients.
Pediatric population
One case of cyst development was reported in an 11-year-old patient and one case of impaired consciousness in a 13-year-old patient; the development of these conditions may be related to treatment with Refacto AF.
The safety of Refacto AF was evaluated in studies involving adults, children, and adolescents who were previously treated (n = 18, aged 12–16 years in the study and n = 49, aged 7–16 years in an additional study), and a trend toward increased frequency of adverse events was observed in children aged 7–16 years compared to adults.
Additional safety data were obtained from studies involving patients who were either previously treated (n = 18 aged under 6 years and n = 19 aged 6–12 years) or previously untreated (n = 23 aged under 6 years). The data indicate that the safety profile is similar to that in adult patients.
Reporting of suspected adverse reactions
It is important to report suspected adverse reactions after marketing authorization. This allows continued monitoring of the benefit-risk balance of the medicinal product. Healthcare professionals should report any suspected adverse reactions in accordance with local requirements.
Shelf life
Presentation – vial: for powder – 3 years, for solvent – 5 years.
Presentation – pre-filled syringe: 3 years.
Storage conditions
Store in the original packaging at 2 to 8 °C. Do not freeze, to avoid damage to the pre-filled syringe.
The product may be stored at temperatures not exceeding 25 °C for up to 3 months within its shelf life. The product cannot be returned to refrigerated storage once stored at room temperature.
The reconstituted solution should be used immediately or within 3 hours after reconstitution (and/or removal of the grey cap from the tip for the pre-filled syringe presentation), provided it is stored at a temperature not exceeding 25 °C.
Keep out of reach and sight of children.
Incompatibilities
Since compatibility studies of this medicinal product have not been conducted, it must not be mixed with other medicinal products, including other infusion solutions.
The infusion set provided in the pack must be used for administration, as factor VIII may adsorb onto the internal surfaces of other infusion equipment.
Packaging
Presentation – vial:
1 vial with lyophilisate, 1 pre-filled syringe with solvent, 1 vial adapter, 1 infusion set, 2 alcohol swabs, 1 plaster, and 1 gauze pad in a cardboard box.
Presentation – pre-filled syringe:
1 pre-filled syringe containing lyophilisate in the upper chamber and solvent (4 ml) in the lower chamber, 1 plunger rod, 1 infusion set, 2 alcohol swabs, 1 plaster, 1 gauze pad, and 1 cap in a cardboard box.
Prescription category Prescription only.
Manufacturer
Wyeth Farma S.A.
Manufacturer's name and address
Autovia del Norte A1, Km 23, desvio Algete, Km. 1, San Sebastian de los Reyes, 28700 Madrid, Spain.