Femoston
Ukraine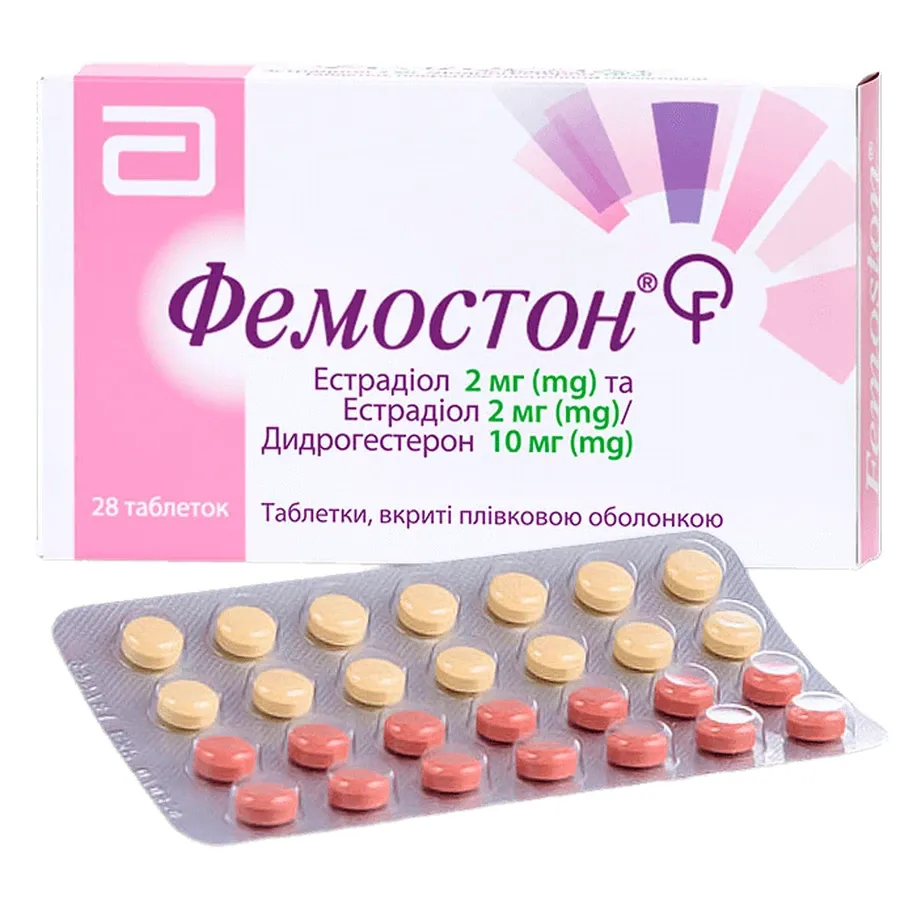
Table of Contents
INSTRUCTION FOR MEDICAL USE OF THE MEDICINAL PRODUCT FEMOSTON® (FEMOSTON®)
Composition:
estradiol tablet
Active substance: estradiol;
1 tablet contains micronized estradiol hemihydrate equivalent to 1 mg or 2 mg of estradiol;
Excipients: lactose monohydrate; hypromellose (HPMC 2910), corn starch, colloidal anhydrous silicon dioxide, magnesium stearate;
Film coating: combined film coating White I (hypromellose (HPMC 2910), polyethylene glycol 400, titanium dioxide (E 171)) – for tablets containing 1 mg of estradiol;
combined film coating Pink I (hypromellose (HPMC 2910), polyethylene glycol 400, talc, iron oxide red (E 172), iron oxide black (E 172), iron oxide yellow (E 172), titanium dioxide (E 171)) – for tablets containing 2 mg of estradiol;
estradiol and dydrogesterone tablet
Active substances: estradiol; dydrogesterone;
1 tablet contains micronized estradiol hemihydrate equivalent to 1 mg or 2 mg of estradiol; micronized dydrogesterone 10 mg;
Excipients: lactose monohydrate; hypromellose (HPMC 2910), corn starch, colloidal anhydrous silicon dioxide, magnesium stearate;
Film coating: combined film coating Grey I (polyethylene glycol 3350, talc (E 553b), polyvinyl alcohol, iron oxide black (E 172), titanium dioxide (E 171)) – for tablets containing 1 mg estradiol and 10 mg dydrogesterone;
combined film coating Yellow II (hypromellose (HPMC 2910), polyethylene glycol 400, talc, iron oxide yellow (E 172), titanium dioxide (E 171)) – for tablets containing 2 mg estradiol and 10 mg dydrogesterone.
Pharmaceutical form. Film-coated tablets.
Main physicochemical characteristics:
tablet containing 1 mg estradiol: round, biconvex, film-coated, white tablet with marking «379» on one side; diameter 7 mm, tablet weight approximately 144 mg;
tablet containing 2 mg estradiol: round, biconvex, film-coated, brick-red tablet with marking «379» on one side; diameter 7 mm, tablet weight approximately 144 mg;
tablet containing 1 mg estradiol and 10 mg dydrogesterone: round, biconvex, film-coated, grey tablet with marking «379» on one side; diameter 7 mm, tablet weight approximately 144 mg;
tablet containing 2 mg estradiol and 10 mg dydrogesterone: round, biconvex, film-coated, yellow tablet with marking «379» on one side; diameter 7 mm, tablet weight approximately 144 mg.
Pharmacotherapeutic group. Medicinal products for urogenital disorders and sex hormones. Combined preparations containing progestogens and estrogens for sequential use. ATC code G03FB08.
Pharmacological properties.
Pharmacodynamics.
Estradiol
The active component, 17ß-estradiol, is chemically and biologically identical to the natural female sex hormone estradiol. It replaces the declining endogenous estrogen production in menopausal women and alleviates menopausal symptoms.
Estrogens prevent bone mass loss following menopause or oophorectomy.
Dydrogesterone
Dydrogesterone is an orally active progestogen with activity comparable to parenterally administered progesterone.
Since estrogens stimulate endometrial growth, in the absence of a progestagen they increase the risk of endometrial hyperplasia and carcinoma. Adding a progestagen to therapy significantly reduces the estrogen-induced risk of endometrial hyperplasia in women with an intact uterus.
Clinical trial data
Reduction of estrogen deficiency symptoms and improvement of bleeding profile.
Reduction of menopausal symptoms was observed within the first few weeks of treatment.
Regular withdrawal bleeding, lasting on average 5 days, occurred in 89% of women treated with Femoston®, estradiol 2 mg + estradiol/dydrogesterone 2 mg/10 mg. Withdrawal bleeding usually started on day 28 of the cycle. Breakthrough uterine bleeding or spotting was reported in 22% of women during the first 3 months of treatment and in 19% of women during months 10–12 of treatment. Amenorrhea (absence of bleeding or spotting) was observed in 12% of cycles during the first year of treatment.
Regular withdrawal bleeding, lasting on average 5 days, occurred in 76% of women treated with Femoston®, estradiol 1 mg + estradiol/dydrogesterone 1 mg/10 mg. Withdrawal bleeding usually started on the last day of the progestagen phase (on average, day 28 of the cycle). Breakthrough uterine bleeding or spotting was reported in approximately 23% of women during the first 3 months of treatment and in 15% of women during months 10–12 of treatment. Amenorrhea (absence of bleeding or spotting) was observed in 21% of cycles during the first year of treatment.
Osteoporosis prevention
Estrogen deficiency after menopause is associated with increased bone resorption and reduced bone mass. The effect of estrogens on bone mineral density is dose-dependent. The protective effect occurs only during treatment. After discontinuation of hormone replacement therapy (HRT), the rate of bone mass loss is similar to that in women who did not receive this therapy.
Data from the Women's Health Initiative (WHI) study and meta-analyses of other trials indicate that HRT, used primarily in healthy women either as estrogen monotherapy or in combination with a progestagen, reduces the risk of hip, vertebral, and other fractures associated with osteoporosis. HRT may also prevent fractures in women with low bone mineral density and/or diagnosed osteoporosis, although data in this population are limited.
After two years of treatment with Femoston®, estradiol 2 mg + estradiol/dydrogesterone 2 mg/10 mg, bone mineral density (BMD) in the lumbar spine increased by 6.7 ± 3.9% (mean ± standard deviation). Lumbar spine BMD increased or remained unchanged in 94.5% of women.
In women treated with Femoston®, estradiol 1 mg + estradiol/dydrogesterone 1 mg/10 mg, lumbar spine BMD increased by 5.2 + 3.8% (mean ± standard deviation). Lumbar spine BMD increased or remained unchanged in 93.0% of women.
Femoston® also affected BMD at the femur.
After two years of treatment with Femoston®, estradiol 2 mg + estradiol/dydrogesterone 2 mg/10 mg, BMD increased by 2.6 ± 5.0% (mean ± standard deviation) at the femoral neck, by 3.5 ± 5.0% (mean ± standard deviation) at the trochanter, and by 4.1 ± 7.4% (mean ± standard deviation) at Ward's triangle. BMD at the three femoral sites increased or remained unchanged in 71–88% of women after treatment with Femoston®, estradiol 2 mg + estradiol/dydrogesterone 2 mg/10 mg.
After two years of treatment with Femoston®, estradiol 1 mg + estradiol/dydrogesterone 1 mg/10 mg, BMD increased by 2.7 ± 4.2% (mean ± standard deviation) at the femoral neck, by 3.5 ± 5.0% (mean ± standard deviation) at the trochanter, and by 2.7 ± 6.7% (mean ± standard deviation) at Ward's triangle. BMD at the three femoral sites increased or remained unchanged in 67–78% of women after treatment with Femoston®, estradiol 1 mg + estradiol/dydrogesterone 1 mg/10 mg.
Pharmacokinetics.
Estradiol
Absorption
Estradiol absorption depends on particle size: micronized estradiol is rapidly absorbed from the gastrointestinal tract.
Table 1 below presents mean steady-state pharmacokinetic parameters of estradiol (E2), estrone (E1), and estrone sulfate (E1S) for each dose of micronized estradiol.
Data are presented as mean (SD).
Table 1
Estradiol 1 mg
| Parameters |
E2 |
E1 |
Parameters |
E1S |
| Cmax (pg/mL) |
71 (36) |
310 (99) |
Cmax (ng/mL) |
9.3 (3.9) |
| Cmin (pg/mL) |
18.6 (9.4) |
114 (50) |
Cmin (ng/mL) |
2.099 (1.340) |
| Cav (pg/mL) |
30.1 (11.0) |
194 (72) |
Cav (ng/mL) |
4.695 (2.350) |
| AUC0-24 (pg·h/mL) |
725 (270) |
4767 (1857) |
AUC0-24 (ng·h/mL) |
112.7 (55.1) |
Estradiol 2 mg
| Parameters |
E2 |
E1 |
Parameters |
E1S |
| Cmax (pg/mL) |
103.7 (48.2) |
622.2 (263.6) |
Cmax (ng/mL) |
25.9 (16.4) |
| Cmin (pg/mL) |
48 (30) |
270 (138) |
Cmin (ng/mL) |
5.7 (5.9) |
| Cav (pg/mL) |
68 (31) |
429 (191) |
Cav (ng/mL) |
13.1 (9.4) |
| AUC0-24 (pg·h/mL) |
1619 (733) |
10209 (4561) |
AUC0-24 (ng·h/mL) |
307.3 (224.1) |
Distribution
Estrogens are present in either unbound or protein-bound form. Approximately 98–99% of an estradiol dose is bound to plasma proteins, of which 30–52% is bound to albumin and about 46–69% to sex hormone-binding globulin (SHBG).
Biotransformation
After oral administration, estradiol undergoes extensive metabolism. The main unconjugated and conjugated metabolites are estrone and estrone sulfate. These metabolites may contribute to estrogenic activity directly or after conversion to estradiol. Estrone sulfate may undergo enterohepatic recirculation.
Elimination
In urine, the main compounds are glucuronides of estrone and estradiol. The elimination half-life ranges from 10 to 16 hours. Estrogens are excreted into breast milk.
Dose- and time-dependence
With daily oral administration of Femoston®, estradiol concentrations reach steady state after approximately five days. In most cases, steady-state concentrations are achieved between days 8 and 11 of treatment.
Dydrogesterone
Absorption
After oral administration, dydrogesterone is rapidly absorbed, with a Tmax of 0.5–1.5 hours.
The following Table 2 presents mean steady-state pharmacokinetic parameters of dydrogesterone (D) and dihydrodydrogesterone (DHD).
Data are presented as mean (SD).
Table 2
Dydrogesterone 10 mg
| Parameters |
D |
DGD |
| Cmax (ng/mL) |
2.54 (1.80) |
62.50 (33.10) |
| Cmin (ng/mL) |
0.13 (0.07) |
3.70 (1.67) |
| Cav (ng/mL) |
0.42 (0.25) |
13.04 (4.77) |
| AUCτ (ng*hr/mL) |
10.17 (5.96) |
312.90 (114.54) |
After administration of a single dose, food delays the time to reach peak plasma concentration of dydrogesterone by approximately 1 hour, resulting in a reduction of peak plasma concentrations of dydrogesterone by about 20%, without affecting the extent of exposure to dydrogesterone and DHD.
Distribution
After oral administration of dydrogesterone, the apparent volume of distribution is large, amounting to approximately 22,000 L. Dydrogesterone and DHD are more than 90% bound to plasma proteins.
Biotransformation
After oral administration, dydrogesterone is rapidly metabolized to form DHD. Plasma levels of the main active metabolite, 20α-dihydrodydrogesterone (DHD), reach peak concentrations at the same time as dydrogesterone. Plasma levels of DHD are substantially higher compared to the parent compound. The AUC and Cmax ratios of DHD to dydrogesterone are approximately 25 and 20, respectively. The mean terminal elimination half-life of dydrogesterone and DHD is about 15 hours. A common feature of all metabolites is the preservation of the 4,6-diene-3-one configuration of the parent compound and the absence of 17α-hydroxylation. This explains the lack of estrogenic and androgenic effects of dydrogesterone.
Excretion
After oral administration of radiolabeled dydrogesterone, on average, 63% of the dose is excreted in urine. The apparent total plasma clearance of dydrogesterone is high, approximately 20 L/min. Complete elimination is achieved within 72 hours. DHD is present in urine predominantly as glucuronide conjugate.
Dose and time dependence
Pharmacokinetics after single and multiple doses are linear over the oral dose range of 2.5 to 20 mg. Comparison of single and multiple dose kinetics shows that the pharmacokinetics of dydrogesterone and DHD do not change upon repeated administration. Steady-state conditions are generally achieved after 3 days of treatment.
Clinical characteristics.
Indications.
Hormone replacement therapy (HRT) for the relief of symptoms associated with estrogen deficiency in menopausal women, not earlier than 6 months after the last menstruation.
Prevention of osteoporosis in postmenopausal women at high risk of fractures. Femoston® should be prescribed to patients only if they are intolerant of or have contraindications to other medicinal products used for osteoporosis prevention (see section "Special precautions").
Experience with treatment in women over 65 years of age is limited.
Contraindications.
- Diagnosed in the past, present, or suspected breast cancer;
- established or suspected estrogen-sensitive tumors (e.g., endometrial cancer);
- established or suspected progestogen-sensitive tumors;
- meningioma or history of meningioma;
- vaginal bleeding of unknown origin;
- untreated endometrial hyperplasia;
- venous thromboembolism, currently present or in history (deep vein thrombosis, pulmonary embolism);
- presence of thrombophilic disorders (e.g., protein C, protein S or antithrombin deficiency; see section "Special precautions");
- acute or recently occurred arterial thromboembolic disease (e.g., angina pectoris, myocardial infarction);
- acute liver disease or history of liver disease if liver function tests have not normalized;
- known hypersensitivity to the active substances or to any of the excipients of the medicinal product;
- porphyria.
Interaction with other medicinal products and other forms of interaction.
Studies on drug interactions have not been conducted.
Estrogen and progestogen effectiveness may be impaired
- Metabolism of estrogens (and progestogens) may be enhanced when co-administered with substances capable of inducing enzymes involved in drug metabolism, particularly CYP450 2B6, 3A4, 3A5, and 3A7. Such substances include anticonvulsants (phenobarbital, carbamazepine, phenytoin) and antibacterial/antiviral agents (e.g., rifampicin, rifabutin, nevirapine, efavirenz).
- Although ritonavir and nelfinavir are known as potent inhibitors of CYP450 3A4, A5, and A7, they actually have an inducing effect when co-administered with steroid hormones.
- Herbal preparations containing St. John's wort (Hypericum perforatum) may also enhance the metabolism of estrogens (and progestogens) via CYP450 3A4.
- Clinically, increased metabolism of estrogens and progestogens may manifest as reduced efficacy and changes in bleeding patterns.
Effect of estrogen-containing HRT on other medicinal products
It has been shown that hormonal contraceptives containing estrogens, when co-administered with lamotrigine, significantly reduce lamotrigine plasma concentrations due to induction of lamotrigine glucuronidation. This may reduce seizure control. Although the potential interaction between hormone replacement therapy and lamotrigine has not been studied, a similar interaction is expected, potentially leading to reduced seizure control in women taking both products simultaneously.
Pharmacodynamic interactions
During clinical trials of hepatitis C virus (HCV) treatment with the combination of ombitasvir/paritaprevir/ritonavir and dasabuvir with or without ribavirin, ALT levels exceeding five times the upper limit of normal were observed significantly more frequently in women using ethinylestradiol-containing medicinal products, such as combined hormonal contraceptives (CHCs). Additionally, with glecaprevir/pibrentasvir or sofosbuvir/velpatasvir/voxilaprevir treatment, increased ALT levels were also observed in women using ethinylestradiol-containing medicinal products such as CHCs. In women who used medicinal products containing estrogens other than ethinylestradiol, such as estradiol, in combination with ombitasvir/paritaprevir/ritonavir and dasabuvir with or without ribavirin, the degree of ALT elevation was similar to that in women not receiving any estrogens; however, due to the limited number of women using other estrogens, caution is advised when co-administering this product with the following combination therapies: ombitasvir/paritaprevir/ritonavir and dasabuvir with or without ribavirin, glecaprevir/pibrentasvir, or sofosbuvir/velpatasvir/voxilaprevir (see section "Special precautions").
Estrogens may interfere with the metabolism of other medicinal products
Estrogens may inhibit CYP450 enzymes involved in drug metabolism through competitive inhibition. This should be particularly considered with medicinal products having a narrow therapeutic index, such as:
- tacrolimus and cyclosporine A (CYP450 3A4, 3A3);
- fentanyl (CYP450 3A4);
- theophylline (CYP450 1A2).
Clinically, this may lead to increased plasma levels of these substances to toxic concentrations. Therefore, careful monitoring of drug levels over an extended period may be required, along with dose reductions of tacrolimus, fentanyl, cyclosporine A, and theophylline.
Special precautions for use.
For the treatment of estrogen deficiency symptoms in women during menopause, hormone replacement therapy (HRT) should be initiated only if these symptoms significantly impair quality of life. A careful benefit-risk assessment of HRT must be performed regularly, at least annually, and treatment should be continued only if benefits outweigh risks.
Data on risks associated with HRT in the treatment of premature menopause are limited. However, due to the low absolute risk in younger women, the benefit-risk ratio in these women is more favorable compared to older women.
Medical examination/follow-up monitoring
Prior to initiating HRT or resuming HRT after a break, a complete medical history (including family history) should be obtained. A physical examination (including gynecological and breast examination) should be performed based on the patient's history, contraindications, and warnings. Regular check-ups are recommended during treatment, with frequency and extent determined individually. Women should be informed about which breast changes should be reported to their physician or nurse (see section below "Breast cancer"). Examinations, including appropriate imaging methods such as mammography, should be performed according to established screening practices, modified according to individual clinical needs.
Conditions requiring patient monitoring
Patients with any of the following conditions currently present, in their history, or with worsening during pregnancy or previous hormonal therapy should be closely monitored. It should be considered that these conditions may recur or worsen during treatment with Femoston®. These include:
- leiomyoma (uterine fibroids) or endometriosis;
- risk factors for thromboembolic disorders (see below);
- risk factors for estrogen-sensitive tumors, e.g., first-degree familial predisposition to breast cancer;
- arterial hypertension;
- liver disease (liver adenoma);
- diabetes mellitus with or without vascular complications;
- gallstone disease;
- migraine or (severe) headache;
- systemic lupus erythematosus;
- history of endometrial hyperplasia (see below);
- epilepsy;
- bronchial asthma;
- otosclerosis.
Meningioma
Cases of meningioma (single and multiple) have been reported during treatment with Femoston®. Patients should be monitored for signs and symptoms of meningioma according to clinical practice. If a patient is diagnosed with meningioma, any treatment with Femoston® should be discontinued (see section "Contraindications"). Tumor regression has been observed after discontinuation of treatment.
Reasons for immediate discontinuation of therapy
Hormone replacement therapy must be immediately discontinued upon identification of any contraindication, as well as in the following situations:
- onset of jaundice or liver function impairment;
- significant increase in blood pressure;
- new onset of migraine-like headache;
- pregnancy.
Endometrial hyperplasia and carcinoma
In women with an intact uterus, the risk of endometrial hyperplasia and carcinoma increases with prolonged estrogen-only HRT. The observed increase in endometrial cancer risk among women taking only estrogen preparations ranges from 2 to 12 times compared to non-users, depending on duration of treatment and estrogen dose (see section "Adverse reactions"). The risk may remain elevated for at least 10 years after discontinuation of treatment.
Cyclic combination of estrogen with a progestogen for at least 12 days per month/in a 28-day cycle, or continuous combined estrogen-progestogen therapy in women with preserved uterus, prevents the excess risk associated with estrogen-only therapy.
Breakthrough uterine bleeding or spotting may occur during the first months of treatment. If bleeding occurs some time after initiation of treatment or persists after discontinuation of therapy, the cause must be investigated, including endometrial biopsy, to exclude malignant neoplasms.
Breast cancer
All available data indicate an increased risk of breast cancer in women taking combined estrogen-progestogen HRT or estrogen-only H0RT. This risk depends on the duration of use.
Combined estrogen-progestogen therapy
Both the randomized placebo-controlled Women’s Health Initiative (WHI) study and meta-analyses of prospective epidemiological studies consistently show an increased risk of breast cancer in women using combined estrogen-progestogen HRT. The increased risk becomes apparent after approximately 3 (1–4) years (see section "Adverse reactions").
Estrogen monotherapy
The WHI study did not show an increased risk of breast cancer in women after hysterectomy who received estrogen-only HRT. Experimental studies have mostly reported a slight increase in risk of breast cancer diagnosis, which is substantially lower than in patients receiving combinations of estrogens and progestogens (see section "Adverse reactions").
Results from a large-scale meta-analysis indicate that after discontinuation of therapy, the increased risk decreases over time, and the time required to return to baseline risk depends on the prior duration of HRT use. In cases where HRT was used for more than 5 years, the risk may persist for 10 years or longer. Hormone replacement therapy, particularly combined estrogen-progestogen therapy, increases mammographic density, which may negatively affect radiological detection of breast cancer.
Ovarian cancer
Ovarian cancer occurs much less frequently than breast cancer. Epidemiological data from a large meta-analysis showed a slightly increased risk in women receiving estrogen-only or combined estrogen-progestogen HRT; this risk emerges within 5 years of use and decreases over time after discontinuation of therapy. Some other studies, including the WHI study, suggest that use of combined HRT may be associated with a similar or slightly lower risk (see section "Adverse reactions").
Venous thromboembolism (VTE)
HRT is associated with a 1.3- to 3-fold increased risk of venous thromboembolism (VTE), i.e., deep vein thrombosis or pulmonary embolism. The occurrence of such events is most likely during the first year of HRT (see section "Adverse reactions").
Patients with known thrombophilic disorders have an increased risk of VTE, and HRT may further increase this risk. Therefore, HRT is contraindicated in this patient group (see section "Contraindications").
Well-established risk factors for VTE include: use of estrogens, advanced age, major surgery, prolonged immobilization, obesity (BMI > 30 kg/m²), pregnancy/postpartum period, systemic lupus erythematosus (SLE), and cancer. There is no consensus on the role of varicose veins in VTE development.
As for all postoperative patients, preventive measures should be taken to avoid VTE after surgery. If prolonged immobilization is expected after elective surgery, HRT should be temporarily discontinued 4–6 weeks before the procedure. Treatment may be resumed only when the woman has fully regained mobility.
Women without a history of VTE but with a first-degree relative who experienced thrombosis at a young age may be offered screening after careful discussion of its limitations (screening detects only a portion of thrombophilic disorders).
If a congenital thrombophilic disorder associated with a family history of thrombosis is identified, or if the disorder is severe (e.g., antithrombin, protein S or protein C deficiency, or a combination of disorders), HRT is contraindicated.
In women already receiving long-term anticoagulant therapy, the benefits and risks of HRT should be carefully weighed.
If VTE develops after starting therapy, the drug must be immediately discontinued. Patients should be warned to seek immediate medical attention if symptoms suggestive of thromboembolism occur (e.g., painful leg swelling, sudden chest pain, dyspnea).
Ischemic heart disease (IHD)
There is no evidence from randomized controlled trials supporting a protective effect against myocardial infarction in women with or without IHD who received combined estrogen-progestogen HRT or estrogen-only HRT.
Combined estrogen-progestogen therapy
The relative risk of IHD is slightly increased with combined estrogen-progestogen HRT. Since the baseline absolute risk of IHD strongly depends on age, the number of additional IHD cases due to estrogen and progestogen use is very small in healthy women close to menopause but increases with age.
Estrogen monotherapy
Data from randomized controlled trials did not show an increased risk of IHD in women after hysterectomy receiving estrogen monotherapy.
Ischemic stroke
Both combined estrogen-progestogen therapy and estrogen monotherapy are associated with a 1.5-fold increased risk of ischemic stroke. The relative risk does not change with age or time since menopause. However, since the baseline absolute risk of stroke strongly depends on age, the overall stroke risk in women receiving HRT increases with age (see section "Adverse reactions").
Other conditions
-
Estrogens may cause fluid retention; therefore, patients with impaired cardiac or renal function should be closely monitored.
-
Women with a previous history of hypertriglyceridemia should be closely monitored during estrogen replacement or hormone replacement therapy, as in rare cases, plasma triglyceride levels have markedly increased during estrogen therapy in these patients, leading to pancreatitis.
-
Exogenous estrogens may induce or exacerbate symptoms of hereditary or acquired angioedema.
-
Estrogens increase levels of thyroxine-binding globulin (TBG), resulting in increased circulating thyroid hormones, measured as protein-bound iodine (PBI), T4 levels (by column or radioimmunoassay), or T3 levels (by radioimmunoassay). Uptake of triiodothyronine (T3) is reduced due to elevated TBG levels. Concentrations of free triiodothyronine (T3) and thyroxine (T4) remain unchanged. Levels of other serum binding proteins, such as corticosteroid-binding globulin (CBG) and sex hormone-binding globulin (SHBG), may increase, leading to increased concentrations of corticosteroids and sex hormones in blood. Concentrations of free and/or biologically active hormones remain unchanged. Concentrations of other plasma proteins (angiotensin-renin substrate, alpha-1-antitrypsin, ceruloplasmin) may also increase.
-
HRT does not improve cognitive function. Some data suggest an increased risk of possible dementia in women who initiate long-term combined or estrogen-only HRT after age 65.
-
Patients with rare hereditary disorders such as galactose intolerance, severe lactase deficiency, or glucose-galactose malabsorption should not take this medication.
-
Femoston® is not a contraceptive.
Elevated ALT levels
In clinical trials involving patients treated for hepatitis C virus (HCV) infection with the combination of ombitasvir/paritaprevir/ritonavir and dasabuvir with or without ribavirin, ALT levels exceeding the upper limit of normal by more than 5 times were observed significantly more frequently in women taking medications containing ethinylestradiol, such as combined hormonal contraceptives (CHCs). Additionally, ALT elevations were also observed in women taking ethinylestradiol-containing medications, such as CHCs, among patients receiving glecaprevir/pibrentasvir or sofosbuvir/velpatasvir/voxilaprevir. In women taking medications containing estrogens other than ethinylestradiol, such as estradiol, in combination with ombitasvir/paritaprevir/ritonavir and dasabuvir with or without ribavirin, the degree of ALT elevation was similar to that in women not receiving any estrogens; however, due to the limited number of women taking other estrogens, caution is advised when co-administering this medication with the following combination therapies: ombitasvir/paritaprevir/ritonavir and dasabuvir with or without ribavirin, glecaprevir/pibrentasvir, or sofosbuvir/velpatasvir/voxilaprevir (see section "Interaction with other medicinal products and other forms of interaction").
Use during pregnancy or breastfeeding.
Pregnancy
Femoston® is not indicated for use during pregnancy. If pregnancy occurs during treatment with Femoston®, the medication should be immediately discontinued.
There are insufficient data on the use of estradiol/dydrogesterone in pregnant women.
Scientific literature has reported that the use of certain progestogens has been associated with an increased risk of hypospadias. However, due to mixed factors during pregnancy, a definitive conclusion on the contribution of progestogens to hypospadias development cannot be made.
Currently, results from most epidemiological studies on accidental fetal exposure to combinations of estrogens and progestogens indicate no teratogenic or toxic risk to the fetus.
Breastfeeding
Femoston® is not indicated for use during breastfeeding.
Effect on fertility.
Femoston® is not indicated for use in women of reproductive age.
Effect on ability to drive and use machines.
Femoston® has no effect or a negligible effect on the ability to drive or operate machinery.
Method of Administration and Dosage
Femoston® is administered orally once daily according to a continuous sequential regimen as described below.
Treatment begins with one tablet containing 1 mg or 2 mg of estradiol taken once daily for the first 14 days of a 28-day cycle.
After this, for the next 14 days, one tablet containing 1 mg or 2 mg of estradiol and 10 mg of dydrogesterone is taken once daily, as indicated on the 28-day calendar pack.
After completion of the 28-day cycle, on day 29, a new 28-day cycle should be started immediately.
Treatment cycles should follow consecutively and continuously.
For the treatment of estrogen deficiency in postmenopausal women, the lowest effective dose should be used as both initial and maintenance therapy, and the duration of treatment should be as short as possible (see also section "Special Warnings and Precautions for Use").
Generally, sequential combined therapy should be initiated with Femoston® containing 1 mg estradiol followed by 1 mg estradiol/10 mg dydrogesterone.
Dosage should be individually adjusted according to clinical response.
In women who are not currently using hormone replacement therapy, or those switching from continuous combined hormone replacement therapy, treatment may be started on any convenient day. In women switching from cyclic or continuous sequential hormone replacement therapy, treatment should begin immediately the day after completion of the previous cycle.
If a tablet has been missed, it is recommended to continue with the next tablet without taking the missed tablet. Missing a tablet may increase the likelihood of breakthrough bleeding or spotting.
Femoston® may be taken regardless of meals.
Children
There is no indication for the use of Femoston® in this patient group.
Overdose
Both estradiol and dydrogesterone are substances with low toxicity. In case of overdose, symptoms such as nausea, vomiting, breast tenderness, dizziness, abdominal pain, somnolence/fatigue, and withdrawal bleeding may occur. It is unlikely that specific or symptomatic treatment will be required in case of overdose.
The above information also applies to overdose in children.
Adverse reactions
The most common adverse reactions in patients treated with estradiol/dydrogesterone during clinical trials were headache, abdominal pain, breast pain/tenderness, and back pain.
Adverse reactions observed during clinical trials (n = 5108) are listed in Table 3, with the frequency specified below:
Table 3
| MedDRA System Organ Classes |
Very common (≥ 1/10) |
Common (≥ 1/100, <1/10) |
Uncommon (≥ 1/1000, < 1/100) |
Rare (≥ 1/10000 to < 1/1000) |
| Infections and infestations |
Vaginal candidiasis |
Cystitis-like syndrome |
||
| Benign, malignant and unspecified neoplasms (incl. cysts and polyps) |
Increased size of fibroids |
|||
| Blood and lymphatic system disorders |
Hemolytic anemia* |
|||
| Immune system disorders |
Hypersensitivity |
|||
| Psychiatric disorders |
Depression, nervousness |
Decreased libido |
||
| Central nervous system disorders |
Headache |
Migraine, dizziness |
Meningioma* |
|
| Eye disorders |
Increased corneal curvature*. Intolerance to contact lenses* |
|||
| Cardiac disorders |
Myocardial infarction |
|||
| Vascular disorders |
Venous thromboembolism**, arterial hypertension, peripheral vascular disorders, varicose veins |
Stroke* |
||
| Gastrointestinal disorders |
Abdominal pain |
Nausea, vomiting, flatulence |
Dyspepsia |
|
| Hepatobiliary disorders |
Liver function disorders (in some cases with jaundice, asthenia or malaise and abdominal pain), gallbladder disorders |
|||
| Skin and subcutaneous tissue disorders |
Allergic skin reactions (e.g. rash, urticaria, pruritus) |
Angioneurotic edema, erythema nodosum*, vasculitis; chloasma or melasma which may persist after discontinuation of treatment* |
||
| Musculoskeletal and connective tissue disorders |
Back pain |
Leg cramps* |
||
| Reproductive system and breast disorders |
Breast pain/tenderness |
Menstrual disorders (including postmenopausal bleeding, metrorrhagia, menorrhagia, oligo-/amenorrhea, irregular menstruation, dysmenorrhea), pelvic pain, cervical discharge |
Increased breast size, premenstrual syndrome (PMS) |
|
| General disorders and administration site conditions |
Asthenic conditions (asthenia, fatigue, malaise), peripheral edema |
|||
| Investigations |
Weight increased |
Weight decreased |
*Adverse reactions reported from spontaneous reports, which were not observed during clinical trials, are included in the frequency category "rare".
** See below for details.
Breast cancer risk
An up to two-fold increased risk of breast cancer diagnosis has been reported in women receiving combined estrogen-progestagen therapy for more than 5 years.
The increased risk in women receiving estrogen-only therapy is lower than in women receiving combined estrogen-progestagen therapy.
The level of risk depends on the duration of use (see section "Special instructions for use").
Below is an estimation of absolute risk based on results from the largest randomized placebo-controlled Women’s Health Initiative (WHI) study and the largest meta-analysis of prospective epidemiological studies.
Table 4
Largest meta-analysis of prospective epidemiological studies
Estimated additional risk of breast cancer after 5 years of use in women with a body mass index of 27 (kg/m²)
| Age at start of HRT (years) |
Number of cases per 1000 women who have never used HRT, over a 5-year period (50-54 years)1 |
Risk ratio |
Number of additional cases per 1000 women using HRT, over 5 years |
| HRT using estrogen-only |
|||
| 50 |
13.3 |
1.2 |
2.7 |
| HRT using combination of estrogen and progestogen |
|||
| 50 |
13.3 |
1.6 |
8.0 |
| Note. Since breast cancer incidence varies between each EU country, the number of additional breast cancer cases will also change proportionally. 1 Based on baseline incidence rates in England in 2015 in women with a body mass index of 27 (kg/m2). |
|||
Table 5
Estimated additional risk of breast cancer after 10 years of use in women with a body mass index of 27 (kg/m2)
| Age at start of HRT (years) |
Number of cases per 1000 women who have never used HRT over a 10-year period (50–59 years)1 |
Relative risk |
Number of additional cases per 1000 women taking HRT over 10 years |
| HRT with estrogen-only therapy |
|||
| 50 |
26.6 |
1.3 |
7.1 |
| HRT with combined estrogen and progestogen therapy |
|||
| 50 |
26.6 |
1.8 |
20.8 |
| 1 Based on baseline incidence rates in England in 2015 for women with a body mass index of 27 (kg/m2). |
|||
Table 6
WHI study in the USA: additional risk of breast cancer after 5 years of use
| Age range (years) |
Number of cases per 1000 women in the placebo group over 5 years |
Relative risk and 95% confidence interval (CI) |
Additional number of cases per 1000 women receiving HRT over 5 years (95% CI) |
| Conjugated equine estrogen (CEE) monotherapy HRT |
|||
| 50–79 |
21 |
0.8 (0.7–1.0) |
|
| CEE + MPA combined estrogen-progestogen HRT‡ |
|||
| 50–79 |
17 |
1.2 (1.0–1.5) |
+4 (0–9) |
| ‡ In a limited analysis involving women who had not used HRT prior to the start of the study, no significant risk was observed during the first 5 years of treatment; after 5 years, the risk was higher than in those who had never used HRT. |
|||
Endometrial cancer risk
Postmenopausal women with intact uterus
The risk of endometrial cancer is approximately 5 cases per 1000 women with intact uterus who do not use HRT.
Estrogen-only HRT is not recommended for women with intact uterus, as it increases the risk of endometrial cancer (see section «Special precautions»). Depending on the duration and dose of estrogen-only therapy, epidemiological studies have reported an increased risk of endometrial cancer ranging from 5 to 55 additional cases diagnosed per 1000 women aged 50 to 65 years.
Adding a progestagen to estrogen-only therapy for at least 12 days per cycle can prevent this increased risk. In the Million Women Study, use of combined (sequential or continuous) HRT for five years was not associated with an increased risk of endometrial cancer (relative risk 1.0 (0.8–1.2)).
Ovarian cancer
Use of HRT containing either estrogen-only or combined estrogen-progestagen HRT has been associated with a slightly increased risk of ovarian cancer (see section «Special precautions»).
Data from a meta-analysis of 52 epidemiological studies reported an increased risk of developing ovarian cancer in women using HRT compared to women who have never used HRT (relative risk 1.43, 95%; confidence interval (CI) 1.31–1.56). In women aged 50 to 54 years who used HRT for 5 years, this resulted in 1 additional case per 2000 women. In women aged 50 to 54 years who did not use HRT, ovarian cancer is diagnosed in approximately 2 per 2000 women over a 5-year period.
Venous thromboembolism risk
HRT is associated with a 1.3- to 3-fold increased relative risk of venous thromboembolism (VTE), i.e. deep vein thrombosis or pulmonary embolism. The occurrence of such events is most likely during the first year of HRT use (see section «Special precautions»). Results from the WHI study are presented below.
Table 7
WHI study: additional VTE risk over 5 years of HRT use
| Age range (years) |
Number of cases per 1000 women in the placebo group over 5 years |
Relative risk and 95% CI |
Additional number of cases per 1000 women taking HRT over 5 years (95% CI) |
| Oral estrogen-only hormone replacement therapy3 |
|||
| 50–59 |
7 |
1.2 (0.6–2.4) |
1 (–3–10) |
| Oral combination estrogen-progestogen therapy |
|||
| 50–59 |
4 |
2.3 (1.2–4.3) |
5 (1–13) |
3 Studies involving women with absent uterus.
Risk of ischemic heart disease
The risk of ischemic heart disease is slightly increased in women receiving combined estrogen-progestogen HRT over the age of 60 years (see section "Special precautions").
Risk of ischemic stroke
The use of estrogen-only therapy and combined estrogen-progestogen HRT is associated with an increased relative risk of ischemic stroke by up to 1.5 times. The risk of hemorrhagic stroke does not increase with HRT use.
The relative risk does not depend on age or duration of treatment; however, since the baseline risk largely depends on age, the overall risk of stroke in women taking HRT increases with age (see section "Special precautions").
Table 8
Pooled data from WHI studies: additional risk of ischemic stroke4 during 5 years of treatment
| Age range (years) |
Number of cases per 1000 women in the placebo group over 5 years |
Risk ratio and 95% CI |
Additional number of cases per 1000 women taking HRT for 5 years (95% CI) |
| 50–59 |
8 |
1.3 (1.1–1.6) |
3 (1–5) |
4 No difference was observed between ischemic and hemorrhagic stroke.
Other adverse reactions reported with estrogen/progestogen therapy (including estradiol/dydrogesterone):
-
Benign, malignant, and unspecified neoplasms: estrogen-dependent neoplasms, both benign and malignant, e.g., endometrial cancer and ovarian cancer; increase in size of progestogen-dependent neoplasms (e.g., meningiomas);
-
Immune system disorders: systemic lupus erythematosus (SLE);
-
Metabolism and nutrition disorders: hypertriglyceridemia;
-
Nervous system disorders: possible dementia, chorea, exacerbation of epilepsy;
-
Vascular disorders: arterial thromboembolism;
-
Gastrointestinal disorders: pancreatitis (in women with pre-existing hypertriglyceridemia);
-
Skin and subcutaneous tissue disorders: erythema multiforme;
-
Renal and urinary disorders: urinary incontinence;
-
Reproductive system and breast disorders: fibrocystic changes in the breasts, cervical erosion;
-
Congenital and genetic disorders: worsening of porphyria;
-
Investigations: increased total thyroid hormone levels.
Reporting of adverse reactions after drug registration is highly important. It enables ongoing monitoring of the benefit-risk balance of the medicinal product. Healthcare and pharmaceutical professionals, as well as patients or their legal representatives, should report all suspected adverse reactions and lack of efficacy through the Automated Pharmacovigilance Information System at the following link: https://aisf.dec.gov.ua.
Shelf life. 3 years.
Storage conditions.
Store in the original packaging at a temperature not exceeding 30 °C. Keep out of reach of children.
Packaging.
Combination pack: 28 tablets in a blister (14 film-coated white tablets of 1 mg + 14 film-coated grey tablets of 1 mg/10 mg in a blister). 1, 2, or 3 blisters per carton.
Combination pack: 28 tablets in a blister (14 film-coated brick-red tablets of 2 mg + 14 film-coated yellow tablets of 2 mg/10 mg in a blister). 1, 2, or 3 blisters per carton.
Prescription category. Prescription only.
Manufacturer. Abbott Biologicals B.V.
Manufacturer's address and place of business.
Veerweg 12, 8121 AA Olst, The Netherlands.